Continuing Education Activity
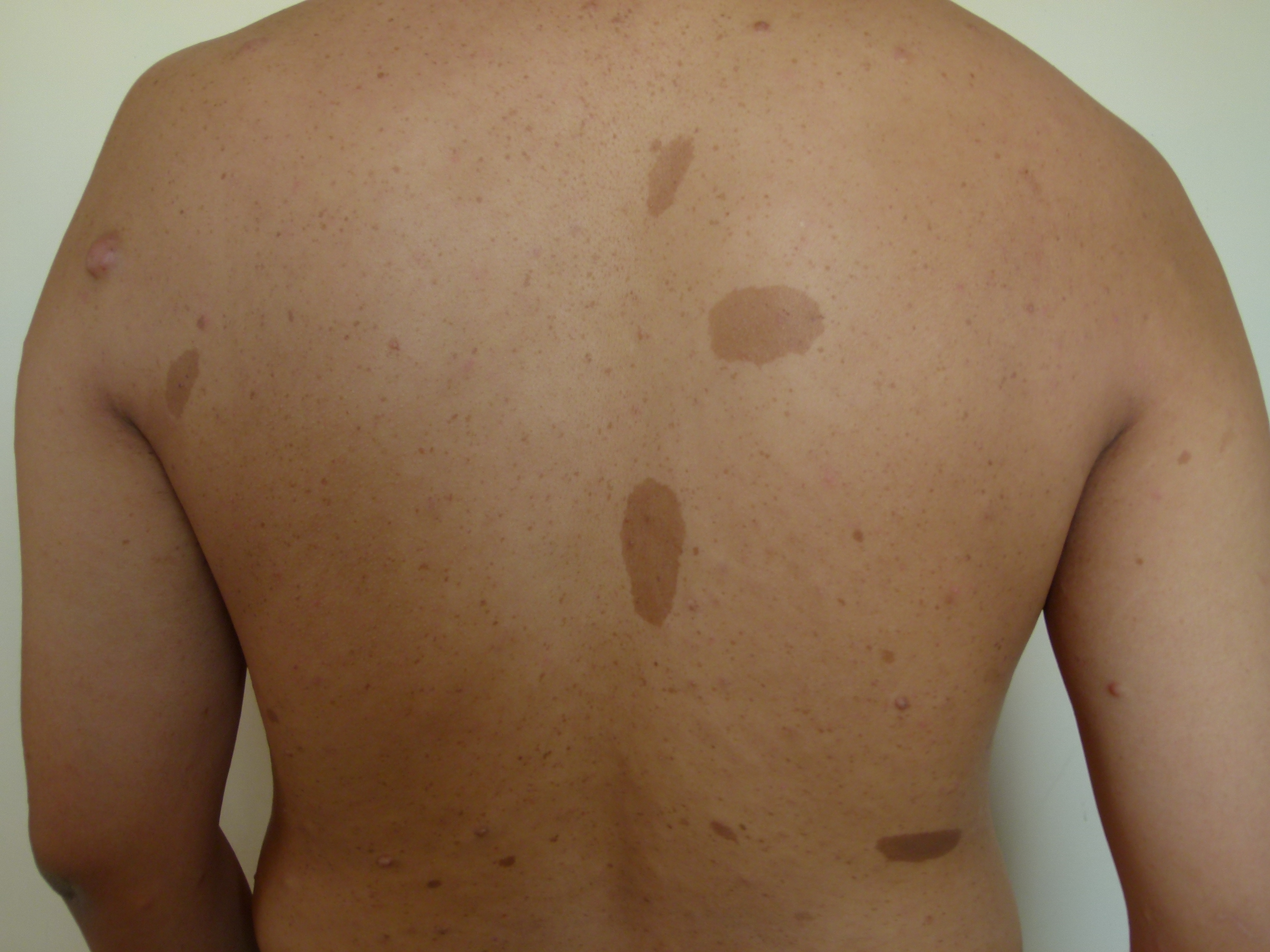
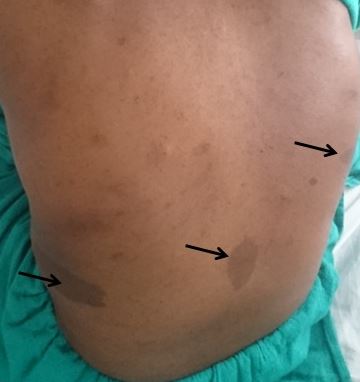
Cafe-au-lait macules (CALMs) are common pigmented lesions found in the general population. They can be described as well-circumscribed, pigmented macules or patches ranging from light to dark brown. Their size may range from a few millimeters to several centimeters (>20cm) and may appear at birth or early life. Multiple CALMs are often associated with genetic syndromes for which the patient and family members need to be evaluated by an interprofessional team. CALMs are mostly benign and are treated with laser therapy for cosmetic purposes. This activity reviews the evaluation and treatment of cafe-au-lait macules and highlights the role of the interprofessional team in evaluating and treating patients with this condition.
Objectives:
- Describe the etiology and associated genetic syndromes with cafe-au-lait macules.
- Summarize the diagnostic criteria for NF1.
- Explain the evaluation of a patient presenting with cafe-au-lait macules.
- Review the importance of collaboration and communication between the interprofessional team and to enhance the delivery of care for patients diagnosed with CALMs associated with genetic syndromes.
Introduction
Cafe-au-lait macules (CALMs) are common hyperpigmented and flat skin lesions found in the general population. They are usually present at birth (congenital) or occur early in life. They may grow in number and size with age. The color varies from light brown to dark brown, and they may be present on any body parts, but the most common location is the trunk and the extremities. The term cafe-au-lait is a French word meaning "coffee with milk."
There are two main types of CALMs. CALMs with regular and clearly demarcated margins ("coast of California"), which is more common. They range in size from a few millimeters to several centimeters (>20cm) and may be present as solitary or multiple spots. The second type of CALM has an irregular margin ("coast of Maine"), which is less common and is usually larger and solitary. The "coast of Maine" pattern is seen in a segmental pigmentary disorder, while the "coast of California" pattern is seen neurofibromatosis 1 (NF1) and related conditions.
CALMs are seen in 95% of patients with NF1. Although solitary CALMs are common in the general population and are familial and inherited as autosomal dominant, multiple spots accompanied by other manifestations may indicate an underlying genetic disorder, and they require further evaluation. A family history of CALMs should be evaluated in any patient with multiple CALMs.[1][2]
Etiology
Cafe-au-lait macules represent a localized area of melanin formation. The exact etiology of CALMs is not known except when associated with genetic syndromes. CALMs are associated with several genetic syndromes like neurofibromatosis (NF1, NF2), McCune-Albright Syndrome, ring chromosome syndromes, constitutional mismatch repair deficiency, tuberous sclerosis, Fanconi anemia, Bloom syndrome, and Silver-Russell syndrome.[1]
CALMs may also serve as a marker for RASopathies (disorders related to RAS mutations). CALMs associated with RASopathies are Legius syndrome, Watson syndrome, and Noonan syndrome with multiple lentigines (formerly known as LEOPARD syndrome).
The etiology of different conditions associated with CALMs include:
- NF1: Due to mutations in the NF1 tumor suppressor gene located on chromosome 17
- NF2: Due to mutations in the NF2 tumor suppressor gene located on chromosome 22
- McCune-Albright Syndrome: Due to a mutation in the GNAS gene
- Legius syndrome: Due to a mutation in the SPRED-1 gene
Epidemiology
Most of the CALMs are present at birth or may appear early in life. CALMs present at birth may be difficult to visualize (Wood's lamp may improve visibility). In patients with NF1, the size and number of CALMs may increase with age. The prevalence of CALMs in newborns varies with race from 0.3% in whites to 18% in African Americans. In school-aged children, the prevalence varies from 13% in whites to 27% in African Americans. CALMs are more frequently seen in black children. While most of the children present with one or two CALMs, three or more CALMs are seen in 1% to 14% of the population.
Pathophysiology
The pathophysiology of CALMs is unclear. However, some studies show that hyperpigmentation is due to an abnormal expression of certain growth factors like stem cell factor and hepatocyte growth factor.
Histopathology
CALMs are caused due to increased melanin in melanocytes and basal keratinocytes. An increase in melanocyte density, an increase in stem cell factor cytokines, and giant melanosomes are noted in NF1 associated cafe au lait macules but are not specific for NF1.
History and Physical
It is important for a clinician to differentiate other pigmented disorders from cafe-au-lait macules. CALMs are mostly seen soon after birth or in early childhood, which may grow in size with time. CALMs may be solitary or multiple, and the margins may be regular or irregular. Solitary CALMs are common in the general population, while the presence of multiple spots requires a detailed evaluation of the patient. Multiple CALMs are often associated with genetic syndromes, most commonly NF1. Thus, a detailed history and physical examination findings in different genetic syndromes associated with multiple CALMs are listed below:
Neurofibromatosis type 1 (NF1)/ von Recklinghausen’s disease
The most frequent disorder seen in association with multiple CALMs is NF1. The estimated population frequency of NF1 is approximately 1 in 3,500. It occurs with equal frequency in males and females and has been identified in all ethnic groups. It occurs due to a mutation in the NF1 gene and follows an autosomal dominant pattern of inheritance. The patient presenting with hyperpigmented skin lesions, difficulty visualizing objects, weakness or neurological deficits, headaches, and pain or deformity in the involved bone require immediate evaluation for NF1.
Diagnostic Criteria: At least two of the following should be present for diagnosing NF1
- Six or more CALMs (greatest diameter>5mm in prepubertal and >15mm in postpubertal individuals)
- One plexiform neurofibroma or two or more neurofibromas of any type
- Axillary or inguinal region freckling (Crowe's sign): pathognomic for NF1
- Optic glioma
- Two or more Lisch nodules (iris hamartomas)
- A distinctive bony lesion, such as sphenoid dysplasia or thickening of the long bone cortex with or without pseudoarthrosis
- A first-degree relative (parent, sibling, or offspring) with NF1 based upon the above criteria[3]
Neurofibromatosis type 2 (NF2)
NF2 is a different disorder from NF1, both clinically and genetically. It occurs due to a mutation in the NF2 gene located on chromosome 22, which produces merlin (tumor suppressor gene) and follows an autosomal dominant pattern of inheritance. Its frequency is approximately 1 in 40,000 births. The clinical features of NF2 include bilateral acoustic schwannomas, cataracts, meningiomas, and ependymomas. The patient may present with a hearing problem, ringing in the ears (tinnitus), and dizziness.[4]
McCune Albright Syndrome
It is a rare genetic condition resulting from a mutation in the GNAS gene affecting G-protein signaling. The classical presentation is a triad of CALMs, fibrous dysplasia of the bone, and precocious puberty. Other endocrinopathies may be present in some patients like hyperthyroidism, hypercortisolism, and hypophosphatemic rickets.[5]
Legius syndrome (NF1-like syndrome)
It is characterized by multiple CALMs, macrocephaly, axillary freckling, lipomas, learning disabilities, and Noonan-like facies. Facial features suggesting Noonan-like facies include hypertelorism (wide-spaced eyes), flat nasal bridge, downward slant of palpebral fissures, thin lips, low-set posteriorly rotated ears, excess nuchal skin, webbed neck, and swollen hands and feet, etc. Patients with Legius syndrome lack neurofibromas and cranial nervous system tumors seen in NF1.[6]
Watson Syndrome
It follows autosomal dominant inheritance and has similar characteristics to NF1. Watson syndrome is characterized by CALMs, pulmonic stenosis, intellectual disability, and short stature.[7]
Noonan syndrome with multiple lentigines (LEOPARD syndrome)
It is an autosomal dominant RASopathy. LEOPARD is an acronym referring to the clinical features which are essential for making a diagnosis. The clinical findings are lentigines, electrocardiogram conduction defects, ocular hypertelorism, pulmonary stenosis, abnormal genitals, retardation of growth, and sensorineural deafness.
Evaluation
CALMs are diagnosed clinically. While evaluating a child with multiple CALMs, it is important to know whether these lesions represent a marker of an underlying systemic disorder. Patients presenting with multiple CALMs (more than 6) should raise suspicion of a genetic syndrome, most commonly NF1. Skin freckling in the inguinal/axillary region and cutaneous neurofibromas can be seen in thorough skin examination. Ophthalmological examination with a slit lamp is equally important to identify Lisch nodules and signs of optic glioma. CALMs are usually distributed in a random pattern, but when found in a single body region, segmental NF1 should be suspected.
Patients with CALMs should be carefully examined for the features of other associated syndromes. Developmental history and progression of the child in school are important for diagnosing syndromes associated with CALMs. Genetic testing can be performed to confirm the diagnosis of suspected genetic syndromes associated with CALMs. For confirmed cases, family members should be evaluated further for findings like multiple CALMs, cutaneous neurofibromas, learning disabilities, skin tumors, spinal cord or brain tumors, etc.
A suspected case of NF2 should be further evaluated with magnetic resonance imaging (MRI) of the brain to know the location of the vestibular schwannoma as well as other tumors associated with NF2 such as meningioma and ependymoma.
A skin biopsy is usually not needed to confirm the diagnosis. On biopsy, an increased amount of melanin in the basal layer along with giant melanosomes can be seen in NF1-associated CALMs.[8]
Treatment / Management
Cafe-au-lait macules have no reported incidence of malignant transformation. Hence, treatment is generally not required unless the patient requests it for improved cosmesis. There has no medical treatment for CALMs until recently. Laser therapy is the mainstay of treatment for these lesions. The lasers that have been used for the treatment of CALMs are Q-switched neodymium:yttrium-aluminum-garnet laser (QSNd:YAG), Q-switched alexandrite laser, Q-switched ruby laser, Copper vapor laser, and Pigmented lesion dye laser. Multiple treatment sessions are required to clear the lesions. The risk associated with laser treatment includes hypopigmentation, transient/permanent hyperpigmentation, scarring, and incomplete clearance.[9]
Patients diagnosed with genetic syndromes are treated with an interprofessional team approach. These patients need to be evaluated by a team that includes a pediatric neurologist, dermatologist, ophthalmologist, geneticist, a specialized nurse, and an orthopedic surgeon. Genetic counseling for the family members and the patient is an important part of the treatment plan for the diagnosed case of CALMs associated with genetic syndromes like NF1 and NF2.[10][11]
Differential Diagnosis
CALMs should be first differentiated from the diffuse causes of hyperpigmentation like Addison's disease, hemochromatosis, and phototoxic allergic reactions. These conditions do not contain the well-demarcated, round borders, as seen in most of the CALMs.
The differential diagnosis of CALMs includes:
- Congenital melanocytic nevus
- Becker nevus
- Nevus spilus
- Lentigo
- Urticaria pigmentosa
- Postinflammatory hyperpigmentation
- Plexiform neurofibroma
- Segmental pigmentation disorder
- Mastocytoma
- Phytophotodermatitis[10]
Prognosis
Cafe-au-lait macules are difficult to treat. Partial clearance may be achieved with laser treatment, but recurrence is common. CALMs are benign lesions with no reported morbidity or mortality, however, associated genetic syndromes may lead to the development of malignant tumors.[9]
Complications
One or two CALMs without any features of genetic syndromes are harmless. CALMs that are multiple and segmental may be a marker for many genetic syndromes. NF1 may lead to the development of malignancy like nerve sheath tumors, optic gliomas, and leukemias.[12]
Tuberous sclerosis may be associated with a cardiac tumor-like rhabdomyoma.[13]
Consultations
Children suspected to have NF1 and NF2 should be evaluated by a team that includes a pediatric neurologist, dermatologist, ophthalmologist, geneticist, and an orthopedic surgeon.
Deterrence and Patient Education
Parents should immediately seek further evaluation if the child has: an undiagnosed pigmented lesion, more than six CALMs, CALMs and nodules on the skin, CALMs and a learning/language/developmental delay, and family members with multiple CALMs or a diagnosis of NF1.
For a patient diagnosed with NF2, annual history and physical examination should be done that includes cutaneous, ophthalmology, and audiology examination. Beginning at 10 years of age, annual brain MRI should be done for early detection of tumors associated with NF2.
Most children diagnosed with NF1 may suffer from learning difficulties, writing problems, and attention difficulties. Parents should be given education about these problems and ways to handle them. Rehabilitation programs should be considered for more serious ones.
Pearls and Other Issues
Legius syndrome can present with similar clinical findings as NF1. It is estimated that 1% to 2% of the patients who meet the diagnostic criteria for NF1 actually have Legius syndrome. Thus, negative results for NF1 molecular testing should prompt a test for Legius syndrome, which is SPRED-1 testing.
Enhancing Healthcare Team Outcomes
Cafe-au-lait macules are benign lesions without any clinical significance, which are common in the general population. Multiple CALMs may be the hallmark for a genetic disorder that needs to be evaluated by an interprofessional team approach. A team consisting of a pediatric neurologist, dermatologist, ophthalmologist, geneticist, orthopedic surgeon, and a specialized nurse should be involved in the care of a patient with a suspected genetic disorder. Annual follow up is required to monitor the progression of the disease.