Continuing Education Activity
Pulmonary hypertension (PH) is defined as mean pulmonary artery pressure greater than 20 mmHg measured during right heart catheterization. The term pulmonary artery hypertension (PAH) describes a subset of patients who also have the presence of pre-capillary hypertension, including an end-expiratory pulmonary artery wedge pressure (less than 15 mmHg) and a pulmonary vascular resistance greater than 3 Woods units. This activity will focus on idiopathic pulmonary artery hypertension (IPAH), which corresponds to sporadic disease, in the absence of family history, and highlight the role of the interprofessional team in its management.
Objectives:
- Describe the evaluation of a patient with pulmonary hypertension.
- Review the presentation of a patient with pulmonary hypertension.
- Outline the treatment and management options available for pulmonary hypertension.
- Describe interprofessional team strategies for improving care coordination and outcomes in patients with pulmonary hypertension.
Introduction
Pulmonary hypertension (PH) is defined as mean pulmonary artery pressure greater than 20 mmHg measured during right heart catheterization. The term pulmonary artery hypertension (PAH) describes a subset of patients who also have the presence of pre-capillary hypertension, including an end-expiratory pulmonary artery wedge pressure (less than 15 mmHg) and a pulmonary vascular resistance greater than 3 Woods units.
PAH has three subgroups: idiopathic, heritable, and pulmonary arterial hypertension related to risk factors or associated conditions. This review will focus on idiopathic pulmonary artery hypertension (IPAH), which corresponds to sporadic disease, in the absence of family history, hereditary forms, or risk factors for PAH.[1][2][3][4]
Etiology
Elevated pulmonary vascular resistance is caused by the proliferation of endothelial and smooth muscle cells, causing tunica media hypertrophy of small-caliber pulmonary arteries. Moreover, the endothelial cells have impaired production of nitric oxide and prostacyclin.
Patients with idiopathic and heritable forms of pulmonary artery hypertension (HPAH) are distinguished by identifying an inheritable genetic mutation, whereas idiopathic forms have an underlying genetic predisposition to develop pulmonary hypertension. Regardless, both forms have genetic mutations that overlap each disease process. IPAH has correlated with the following genetic mutations: bone morphogenetic protein receptor type 2 (BMPR2), mothers against decapentaplegic, Drosophila, a homolog of 1 and 9 (SMAD 1 and 9), potassium channel subfamily K member 3 (KCNK3), and caveolin 1 (CAV1). While a genetic basis exists in the development of IPAH, there are environmental factors that likely contribute to the pathogenesis. For instance, in a review of 340 patients with idiopathic PAH, it was found that these patients were 10.14 times more likely to have a history of stimulant use (amphetamine, methamphetamine, cocaine) in comparison to patients with pulmonary artery hypertension and a risk factor (familial disease, collagen vascular disease, congenital systemic-to-pulmonic shunts, or use of fenfluramine/dexfenfluramine). Further research is needed to elucidate these findings fully.
Epidemiology
In a US registry of 2039 patients with pulmonary artery hypertension, the average age was 51.7 (plus or minus 14.5 years) with a female-to-male ratio of 3.9:1.0. Of those patients, 46.6% (950) were classified as IPAH. Estimates are that IPAH affects 1 in 1 million, usually young females who are otherwise normal. The median survival, if left untreated, is 2.8 years.[5][6]
Pathophysiology
The initial insult to the pulmonary arteries in idiopathic pulmonary hypertension is likely a combination of genetic predisposition with environmental influences. With that said, the pathophysiology of the development of pulmonary artery hypertension is the same except for different biomarkers that lead to a cascade of events, ultimately causing vascular remodeling of the vessel wall; this is an important distinction because knowing the underlying cellular mediators help guide targeted therapies. The vascular remodeling process innate to all forms of pulmonary artery hypertension begins with excessive vasoconstriction, which is related to abnormal expression/function of potassium channels. This leads to a decrease in vasodilators (nitric oxide, prostacyclin) and an increase in vasoconstrictors (endothelin-1).
The decreased lumen size of the pulmonary vasculature causes increased right ventricular afterload. In turn, right ventricular dilation results in septal flattening and Doppler notching and ultimately increased pulmonary artery systolic pressure. To estimate the PASP, continuous-wave Doppler will determine the peak tricuspid regurgitant jet velocity (TRV). From here, the modified Bernoulli equation is applied (4v^2) and added to the right atrial pressure to estimate PASP. If PASP is greater than 50 mmHg, then pulmonary hypertension is very likely, and the patient should obtain a referral for a right heart catheterization.
Histopathology
In the presence of chronically elevated pulmonary artery pressure, progressive histological changes occur in the pulmonary vasculature. In the pulmonary arteries and arterioles, there is medial hypertrophy, marked intimal fibrosis, and thickening of the adventitial layer with occasional plexiform lesions. The intimal lesions account for the most deleterious effects on reducing the lumen of the pulmonary artery and typically comprise of fibrotic, plexiform, and concentric lesions. The concentric lesions are organized myofibroblasts or endothelial cells that form “onion-skin” layers seen in severe forms of pulmonary hypertension.
History and Physical
Initial symptoms include dyspnea, fatigue, chest pain, near syncope/syncope, and lower extremity edema. These vague symptoms likely contribute to causing a delay in diagnosis by an average of 2 years for most patients. Physical exam findings in a typical patient with pulmonary hypertension may include an increased pulmonic component of the second heart sound (P2) (93%), tricuspid regurgitation murmur (40%), pulmonic insufficiency murmur (13%), and peripheral edema (32%).
Evaluation
By definition, to diagnose idiopathic pulmonary artery hypertension, all other causes of elevated pulmonary artery pressures must be ruled out. A clinician should start with the least invasive testing before pursuing the right heart catheterization, which cinches the diagnosis of pulmonary artery hypertension. Serological testing should focus on ruling out connective tissue disease (scleroderma, systemic sclerosis, lupus), HIV, cirrhotic liver disease causing pulmonary hypertension, and schistosomiasis.[7][8][9]
Electrocardiogram (ECG), chest radiography, and pulmonary function tests may provide important clues to diagnosing pulmonary hypertension, though none of these modalities provide enough sensitivity or specificities to serve as a screening tool. On ECG, right-axis deviation and right ventricular hypertrophy are observable 79% and 87% of the time. Chest radiographic findings of increased pulmonary vascular pressure include attenuated vascular markings and enlargement of the main and hilar pulmonary arteries. Patients with idiopathic pulmonary arterial hypertension may have mild reductions in total lung capacity (TLC) and forced vital capacity (FVC). The diffusing capacity of carbon monoxide (DLco) is also significantly less in patients with IPAH. Measurement of oxygen saturation during sleep and exercise must be part of the workup for patients with suspected pulmonary hypertension, as the slightest degree of hypoxia may induce further pulmonary artery vasoconstriction propagating hypertension.
The echo-doppler is the first test to obtain on patients with suspected elevated pulmonary artery pressure. The echo-doppler is useful as a screening tool; however, it is only 50% accurate in estimating PASP. The variability of determining PASP on echo-doppler is due to dynamic changes in volume status of a patient (causing changes in vena cava diameter, therefore different estimates of right atrial pressure), doppler probe positioning, and variability in estimating peak jet velocity. Estimating PASP with Doppler echocardiography to make a diagnosis of pulmonary hypertension should be in conjunction with an overall Doppler examination of the heart as some patients may have underestimated PASP yet have right ventricular dilation or dysfunction. The role of exercise echocardiography in identifying or prognosticating patients with PAH remains controversial.
A right heart catheterization is necessary to diagnose pulmonary artery hypertension (as opposed to pulmonary hypertension). This intervention gives a complete hemodynamic assessment, including measurement of pulmonary artery pressure, pulmonary capillary wedge pressure, pulmonary vascular resistance, and transpulmonary and diastolic pressure gradients. Left heart catheterization is often pursued in the same setting because left heart filling pressures may lead to an alternate cause of elevated pulmonary artery pressures (left heart failure causing pulmonary venous congestion).
The fifth world symposium on PAH defines pulmonary hypertension as a mean PAP greater than 20 mmHg at rest. Pulmonary artery hypertension is defined by mean PAP greater than 20 mmHg at rest and pulmonary capillary wedge pressure (PCWP) less than 15 mmHg, and peripheral vascular resistance (PVR) greater than 3 Woods units.
Treatment / Management
Before initiating therapy, patients with IPAH should undergo vasoreactivity testing to determine if the patient would have a response to calcium channel blockers (CCB). The test involves administrating a vasodilator (nitric oxide, epoprostenol, adenosine) during right heart catheterization and is considered positive if the mean pulmonary artery pressure decreases by 10 mmHg and to a value less than 40 mmHg. Additionally, the patient’s cardiac output should increase or remain unchanged and without a drop in systemic blood pressure. These patients may benefit from calcium channel blockers such as dihydropyridine, nifedipine, or diltiazem.
For patients classified as WHO Class II, endothelin-blocking antagonists such as ambrisentan or bosentan, in addition to phosphodiesterase type 5 inhibitors sildenafil, are recommended (strength A recommendation). For WHO Class III, in addition to ambrisentan or bosentan plus sildenafil, intravenous (IV) epoprostenol or inhaled iloprost can be used (strength A recommendation). Epoprostenol IV and inhaled iloprost are both prostacyclin analogs causing dilation of pulmonary vessels, inhibiting platelet aggregation, and smooth muscle proliferation. If there is an inadequate clinical response, therapeutic alternative for prostacyclin analog for WHO Class III patients include subcutaneous treprostinil (strength B recommendation). Tadalafil may also be an option if the patient does not tolerate sildenafil.
For patients classified in WHO Class IV, intravenous epoprostenol is the mainstay therapy. If an inadequate clinical response remains, combination therapy, including prostanoids, a phosphodiesterase inhibitor, and endothelin-blocking antagonist, merits consideration. Further options for end-stage or rapidly progressive IPAH patients include atrial septostomy and/or lung transplant. Other adjunctive therapies to be considered on a case-by-case basis include oxygen and diuretics to maintain euvolemia.
Differential Diagnosis
Secondary causes of elevated pulmonary artery pressures must be ruled out to diagnose idiopathic pulmonary hypertension. Therefore, the differential diagnosis of idiopathic pulmonary hypertension includes congenital or acquired heart disease, veno-occlusive disease, pulmonary diseases, chronic hypoventilation secondary to sleep apnea, chronic exposure to high altitude, liver disease, ingestion of anorectic medications or pharmaceutical agents, and fibrosing mediastinitis.
Staging
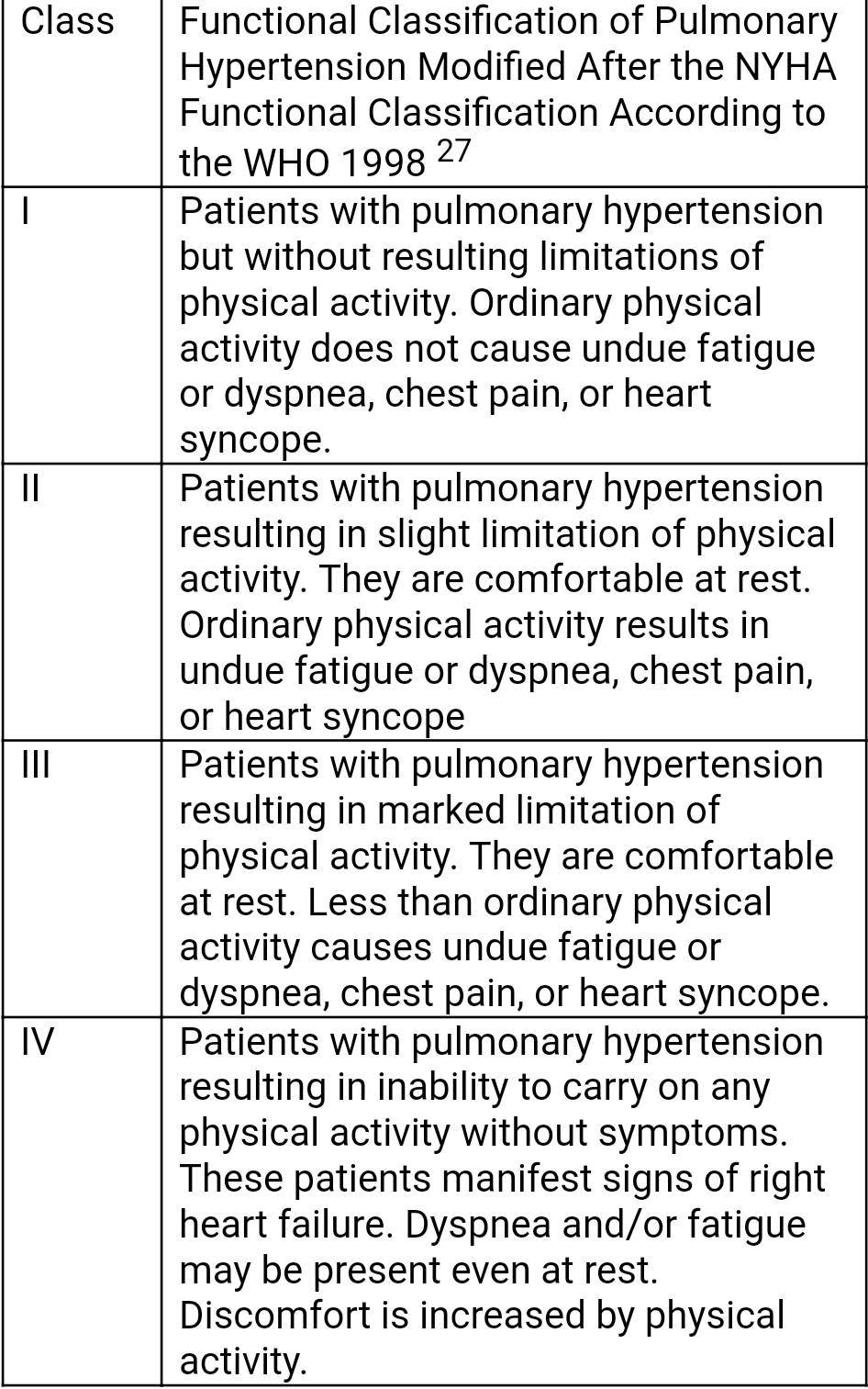
Patients must undergo exercise capacity testing to determine the severity of disease and assess their response to treatment. Therefore, before beginning therapy, all patients should have a baseline exercise assessment done. Objective measurement is the 6-minute walk test, which is an independent predictor of mortality and can assess response to therapy. From exercise capacity, a patient’s functional class can be determined as per WHO Functional Classification for Pulmonary Hypertension.
Prognosis
The REVEAL Registry PAH Risk Score Calculator is a tool clinicians may use to help prognosticate patients with pulmonary artery hypertension.
The tool has validation as a predictive algorithm for 1-year survival. Factors that are independently associated with decreased survival include the following:
- Men older than 60 years
- PAH associated with portal hypertension or connective tissue disorder
- Family history of PAH
- WHO Class III or IV renal insufficiency
- Resting systolic BP less than 110 mmHg
- Heart rate greater than 92 beats per minute
- Six-minute walk test less than 165 m
- Brain natriuretic peptide greater than 180 pg/ml
- Pulmonary vascular resistance greater than 32 Woods units
- Presence of pericardial effusion on echocardiogram
- TAPSE (tricuspid annular plane systolic excursion) less than 1.5 cm, percentage predicted diffusing capacity of the lung for carbon monoxide (DLCO less than 32%)
Complications
Pulmonary artery hypertension, as well as pulmonary hypertension, can cause a variety of complications. Beginning with vascular complications, patients can have pulmonary artery dilatation secondary to the increased volume and shearing stress on the vessel wall. The potential effects of a dilated pulmonary artery include extrinsic compression of the left main stem coronary artery.
A rare complication is pulmonary artery dissection, usually diagnosed in post-mortem exams. While rare, it is associated with congenital heart disease and idiopathic pulmonary artery hypertension.
Chronic pulmonary artery hypertension leads to increased pulmonary artery resistance, which puts increased stress on the right ventricle' this causes the right ventricle to hypertrophy and then dilate to accommodate the increased preload. Should these mechanisms fail, then tricuspid regurgitation, pericardial effusion, and potentially cardiac cirrhosis can develop.
Iatrogenic causes of complications from pulmonary artery hypertension come from line infections in patients being treated with prostacyclin analogs. Administration of this drug must be via a central line, and due to the chronic nature of the disease, the line becomes a nidus for infection.
Postoperative and Rehabilitation Care
Patients with pulmonary hypertension are universally at higher risk during surgery; this is dependent on the severity and co-morbid conditions and differs from patient to patient. Specific risks during the peri-operative period include hemodynamic instability and right heart failure. The mechanism of right heart failure is increased pulmonary vascular resistance resulting in decreased cardiac output. Minimizing pulmonary vascular resistance peri-operatively is of central importance in preventing right ventricular failure. To date, there has not been data to support the treatment of right ventricular failure due to increased pulmonary vascular resistance. Pressor support has its limitations, as increasing peripheral vascular resistance may also increase pulmonary vascular resistance. A combination of inhaled iloprost or intravenous milrinone with oral sildenafil may reduce pulmonary vascular resistance without decreasing systemic blood pressure. Another option is using low-dose vasopressin to augment systemic vascular resistance and lower pulmonary vascular resistance through the release of nitric oxide from the pulmonary vessel endothelium.
Deterrence and Patient Education
All patients with idiopathic pulmonary hypertension require supportive therapy, including infection prevention (age-appropriate vaccinations), supervised rehabilitation, and psychological/social support. Female patients of childbearing age should be counseled on birth control as pregnancy could have detrimental effects on the patient.
Pearls and Other Issues
The term pulmonary artery hypertension has three subgroups: idiopathic, heritable, and pulmonary arterial hypertension related to risk factors or associated conditions. In the pulmonary arteries and arterioles, there is medial hypertrophy, marked intimal fibrosis, and thickening of the adventitial layer with occasional plexiform lesions. The decreased lumen size of the pulmonary vasculature causes increased right ventricular afterload. In turn, right ventricular dilation results in septal flattening and ultimately increased pulmonary artery systolic pressure.
The echo-doppler is the first test to obtain on patients with suspected elevated pulmonary artery pressure. If a patient is found to have a mean PAP greater than 20 mmHg at rest and pulmonary capillary wedge pressure (PCWP) less than 15 mmHg and peripheral vascular resistance (PVR) greater than 3 Woods units on right heart catheterization, then pulmonary artery hypertension can be diagnosed. All other causes of elevated pulmonary artery pressure must be ruled out to diagnose idiopathic pulmonary artery hypertension. The severity of the disease is measurable by a patient's ability to complete a 6-minute walk test and is scaled according to the NYHA Functional Classification. Before initiating therapy, patients with IPAH should undergo vasoreactivity testing to determine if the patient would have a response to calcium channel blockers (CCB). The mainstay treatment for IPAH includes endothelin-blocking antagonists, prostanoids, and phosphodiesterase inhibitors.
Enhancing Healthcare Team Outcomes
Idiopathic pulmonary hypertension is a life-threatening disorder that significantly affects the quality of life and, over time, leads to right heart failure. A timely evaluation, proper treatment, regular follow-up, and patient education can positively affect the outcome of the disease. For this, integrated, interprofessional care with the involvement of different subspecialties is required. The physician, along with a team of nurse specialists, pharmacists, and social workers, functioning as an interprofessional team, should coordinate closely to provide the best possible care and counsel these patients.
Non-specific symptoms of the disease usually result in a delay in diagnosis. The family physician suspects the disease based on non-invasive tests (echocardiogram, X-ray). Once the presumptive diagnosis of pulmonary hypertension has been made, it is very critical to classify the type of pulmonary hypertension as the treatment for one group may cause deleterious effects if used for another group. These patients are, therefore, referred to different subspecialty physicians to determine the cause. A referral to the pulmonologist is essential for sleep studies and pulmonary function tests to exclude lung disease as a cause. To rule out left heart disease, patients are referred to a cardiologist. Right heart catheterization to confirm the diagnosis is also done by the cardiologist. Similarly, to rule out other causes, the patients are also referred to rheumatologists and other doctors. It is also important to counsel the patients so that they can live with this disease. The pharmacist should examine all current medications, as well as verify the dosing and interactions of any new medications prescribed for the condition, and report back to the team should any issues arise.
Treatment of the cause of pulmonary hypertension is the key to managing the disease. Hence, collaboration with physicians, physician assistants, and nurse practitioners in different specialties, working as a cohesive interprofessional team, is essential to manage pulmonary hypertension.[10][11][12] [Level 5]