Continuing Education Activity
Phenylketonuria (PKU) is an inborn error of metabolism (IEM) most often caused by missense mutations in the gene encoding phenylalanine hydroxylase (PAH), which catalyzes the hydroxylation of phenylalanine (Phe) to generate tyrosine (Tyr). PKU belongs to a class of aminoacidopathies termed toxic accumulation IEMs, in which the accumulation of an amino acid or its metabolite is toxic. Elevated blood Phe levels and decreased Tyr levels characterize PKU. Newborns with PKU can appear normal at birth with the first signs appearing after several months. These signs can include musty odor from skin and urine, fair skin, eczema, seizures, tremors, and hyperactivity. This activity examines the presentation, evaluation, and management of phenylketonuria and stresses the role of an interprofessional team approach to the care of affected patients.
Objectives:
- Explain how to counsel a patient and their family about lifestyle to decrease the risk of progression and complications of phenylketonuria.
- Describe the history and physical exam findings typically seen in patients with phenylketonuria.
- Explain the proper management of phenylketonuria.
- Describe how coordination of the interprofessional team can lead to more rapid diagnosis of phenylketonuria and subsequently decrease associated morbidity in affected patients.
Introduction
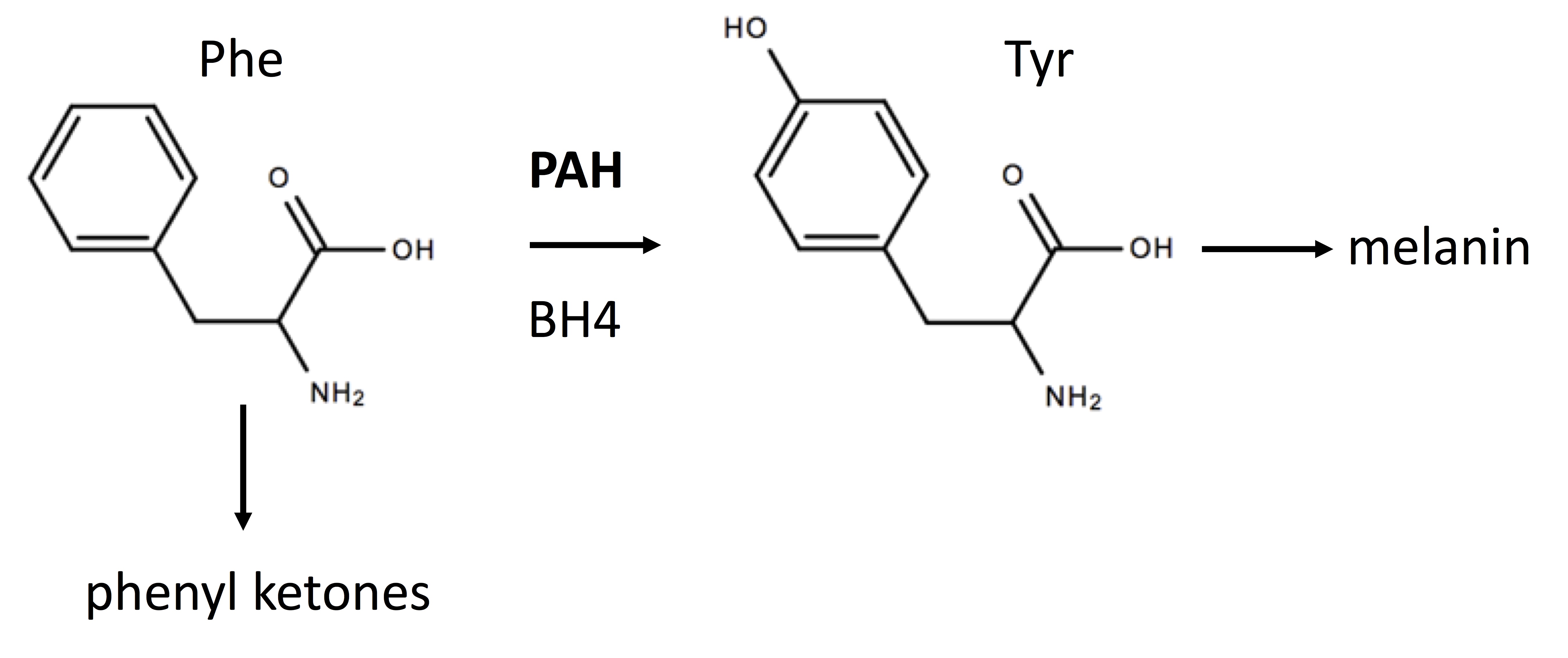
Phenylketonuria (PKU) is an inborn error of metabolism (IEM) most often caused by missense mutations in the gene encoding phenylalanine hydroxylase (PAH) which catalyzes (see Figure 1) the hydroxylation of phenylalanine (Phe) generating tyrosine (Tyr).[1] PKU belongs to a class of amino acid aminoacidopathies termed “toxic accumulation-IEMs” where the circulating toxin is an amino acid or its metabolites. Mutations in an enzyme, such as PAH, are recessive since one functioning enzyme with the wild-type allele is sufficient.
Tetrahydrobiopterin (BH4) binds to the catalytic domain of PAH and is a cofactor for this reaction. See figure. PAH is primarily a hepatic enzyme. Elevated blood Phe levels and decreased Tyr levels characterize PKU. Newborns with PKU can appear normal at birth with the first signs appearing after several months. These signs can include musty odor from skin and urine, fair skin, eczema, seizures, tremors, and hyperactivity.
Etiology
There are over 1000 mutations resulting in PKU, the most common replaces arginine (Arg) with tryptophan (Trp) at position 408 (i.e., Arg408Trp). Mutations in PAH causing PKU are generally due to a reduced PAH activity or a reduced PAH expression resulting in increased blood levels of Phe and decreased Tyr levels. Most PKU-causing PAH variants result from PAH misfolding and/or instability.[2] The Arg408Trp mutation results in “classic” PKU where blood Phe levels are greater than 1200 micromolar. Less severe mutations result in mild PKU, with blood Phe levels from 600 to 1200 micromolar, and mild hyperphenylalaninemia with blood Phe levels being less than 600 micromolar. Phe is an essential amino acid, and its conversion to Tyr is normally well-regulated to provide sufficient levels for protein synthesis while maintaining levels sufficiently low to be nontoxic.[2]
Most PKU patients have 2 different PKU variants, i.e., they are compound heterozygotes.[3] PKU patients can, for example, have 2 severe alleles (severe/severe) or 1 severe allele and 1 mild allele (severe/mild). It is possible that some mild PAH gene variants code for an enzyme with reduced affinity for BH4 (a Km variant) and/or an enzyme variant with increased stability and half-life as a resulting of BH4 binding. Some 49% of PKU patients respond to oral BH4 supplementation with a 30% decrease in blood Phe levels despite having normal levels of BH4.[3]
Epidemiology
The overall incidence of PKU in the United States is about 1/15,000. This incidence is greater for Caucasian and Native American populations and less for African American, Hispanic and Asian populations. There is a large worldwide variation in PKU incidence. In Turkey, the incidence is particularly high with 1/4000 live births.
Pathophysiology
The pathophysiology of PKU is primarily attributed to elevated levels of Phe and its metabolites such as the keto acid, phenylpyruvate. Decreased levels of Tyr may also play an adverse role since this amino acid is an important precursor of 3 catecholamine neurotransmitters: dopamine, norepinephrine, and adrenaline. Tyr is also a precursor of the skin pigment, melanin. Reduced Tyr can cause decreased melanin synthesis and light skin and hair. The precise molecular pathophysiological mechanisms giving rise to cognitive impairment in PKU are not fully understood.[1] Increased oxidative stress has emerged as a possible underlying mechanism for the neurodegeneration observed in PKU. Other mechanisms for PKU pathophysiology include altered neurotransmitter metabolism, decreased cerebral protein synthesis, and energetics.
History and Physical
In the United States, PKU is usually detected at birth by newborn screening tests and dietary therapy started in consultation with a dietitian and geneticist/metabolism specialist. Mild forms of PKU in a newborn can, however, go undetected if the mother is discharged too soon or if the newborn has not consumed any protein. Signs and symptoms of the disease that become more pronounced when untreated include delayed developmental milestones, microcephaly, hypopigmentation, hyperactivity/behavior problems, seizures, and a musty odor to skin and urine. However, affected children who are diagnosed and appropriately treated at birth are less likely to show symptoms. Long-term care focuses on adherence to dietary therapy, monitoring of blood Phe and Tyr levels and assessing for potential cognitive impairments.
Evaluation
PKU in the United States is detected by the state newborn screening program which measures the Phe/Tyr molar ratio on a filter paper blood spot (from a heal prick) by tandem mass spectrometry (MS/MS). This test is usually done 1 or 2 days after birth. Newborns with PKU can appear normal at birth with the first signs appearing after several months. Neonates with elevated blood Phe may have a BH4 deficiency which could also result in elevated Phe levels. Commercial microarray genotyping (for example, 23andme.com) for single nucleotide polymorphisms (SNPs) can provide the carrier status for PKU variants common in those with Northern European descent. Knowing carrier status could be useful for future parents; if both are carriers of a PKU variant, there is a 25% of having a child with PKU.
Treatment / Management
When diagnosed early, classic PKU can be treated by life-long dietary therapy focused on maintaining low Phe levels and adequate Tyr intake. These dietary interventions are generally effective at preventing the most severe cognitive impairment due to high Phe levels. Nevertheless, dietary therapy for PKU has been associated with deficiencies in selenium, copper, magnesium, and zinc.
The management of PKU is complex and dietary non-adherence often increases in adolescence and early adulthood, particularly due to social issues. Besides a low Phe diet, promising drugs are being developed for PKU. Oral sapropterin dihydrochloride (KUVAN), a synthetic form of BH4, could help lower Phe levels in some PKU patients. It is not yet possible to predict which PKU patients are BH4 responders, but a 30-day trial can be undertaken to make this determination. Encouragingly, sapropterin dihydrochloride treatment improves brain function in some PKU patients.[4]
Enzyme replacement therapy for PKU has not been possible since PAH is unstable. The FDA has recently approved an enzyme substitution therapy for PKU. In this approach, a “substitution” enzyme is delivered to the PKU patient that can lower Phe levels. This substitution enzyme is a PEGylated phenylalanine ammonia lyase (called pegvaliase) that degrades Phe. Pegvaliase is approved only for adult patients having uncontrolled phenylalanine levels. It is not yet clear if pegvaliase will permit a less strict dietary regimen or be of long-range benefit in preventing or reversing cognitive impairment.
Women of childbearing age with PKU should receive counseling concerning the benefits of strict dietary therapy both before and during pregnancy. Elevated maternal Phe levels during pregnancy may cause fetal brain damage and congenital heart disease.[5] The adverse fetal effects of elevated Phe during pregnancy can occur whether or not the fetus has a PKU variant.
Treatment of IEMs has traditionally relied on treating the nutritional and/or metabolic environment, for example, replacement enzyme therapy, rather than the genetic disorder itself. Rapid advances in gene therapy and its safety may one day be possible.[6] As of 2018, phase 1, gene-therapy trials for PKU remain in the planning stage.
Differential Diagnosis
As shown in the figure below, the conversion of Phe to Tyr by PAH requires BH4 and a deficiency of BH4 can cause hyperphenylalaninemia (HPA) even in the presence of wild-type PAH. If a newborn has a positive screen for HPA, further testing should be performed for pterins. Although beyond the scope of this review, it is possible to diagnose all forms of BH4 deficiencies based on the pattern of pterins.
Prognosis
A recent review shows that some cognitive impairments can occur even with early PKU treatment with deficits noted in executive functions.[7]
Consultations
Effective management of PKU is best accomplished with a team approach. Depending upon the severity, this typically involves a primary care physician, a dietitian with PKU experience and a genetic counselor or geneticist/metabolism specialist. A clinical psychologist and social worker, as well as a developmental pediatrician, may be needed to address social and developmental concerns. Women of childbearing age with PKU and considering pregnancy, an obstetrician-gynecologist should be consulted before an anticipated pregnancy.
Deterrence and Patient Education
Medical centers specializing in IEMs often have sufficient resources to provide both a team approach for PKU and extensive patient education programs.
Enhancing Healthcare Team Outcomes
PKU is a rare, metabolic disorder that is often detected at birth. Because of its high morbidity, healthcare workers including nurse practitioners must be aware of the screening test available to identify this disorder. In the United States, PKU is usually detected at birth by newborn screening tests and dietary therapy started in consultation with a dietitian and geneticist/metabolism specialist. Mild forms of PKU in a newborn can, however, go undetected if the mother is discharged too soon or if the newborn has not consumed any protein. The outcomes for infants treated at birth are good, but recent evidence suggests that a significant number of children may still have residual cognitive deficits despite early treatment.