Introduction
Aldosterone is a mineralocorticoid hormone produced in the zona glomerulosa of the adrenal cortex that influences water and salt regulation in the body. Aldosterone's primary function is to act on the late distal tubule and collecting duct of nephrons in the kidney, favoring sodium and water reabsorption and potassium excretion while also contributing to acid-base balance. To execute these tasks, it influences epithelial sodium channels, sodium-potassium exchange pumps, hydrogen ion ATPases, and bicarbonate-chloride antiporters.[1] Aldosterone affects blood pressure by regulating the sodium gradient in the nephron to either increase or decrease the water reabsorped to contribute to the volume of the extracellular fluid (ECF). This, however, is not to be confused with the effect of anti-diuretic hormone (ADH), also referred to as vasopressin. ADH is often released simultaneously with aldosterone in order to support water reabsorption to the ECF by mobilizing aquaporin channels to the apical (lumen-facing) membrane of principal cells in the collecting tubule. Overall, aldosterone is a key player in the multi-factorial regulation of salt, potassium, blood pressure, and acid-base balance.[2][3][4]
Cellular Level
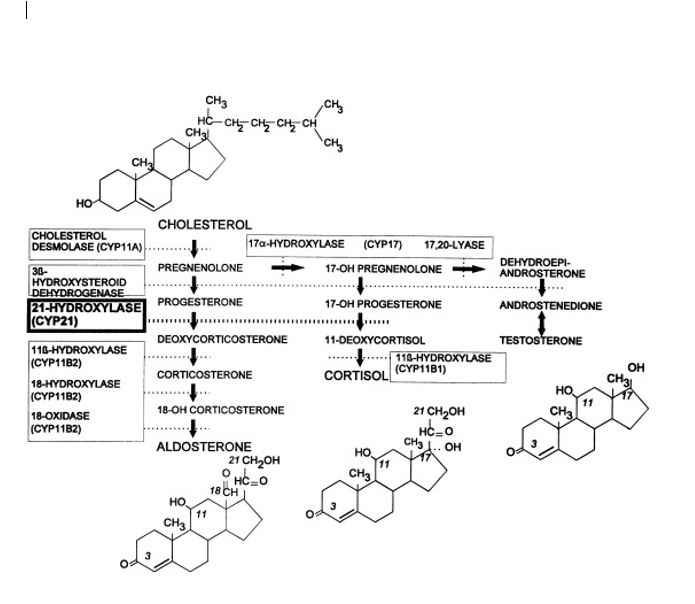
Aldosterone is created from cholesterol within the zona glomerulosa of the adrenal glands. Cholesterol interacts sequentially with the enzymes 3-beta-hydroxysteroid dehydrogenase, 21-alpha-hydroxylase, 11-beta-hydroxylase, and steroid 18-hydroxylase (also called aldosterone synthase) to produce 11-beta, 21-dihydroxy-3, 20-dioxopregn-4-en-18-al (aldosterone). These enzymes also function in the production of other steroid hormones from cholesterol in the adrenal glands, including glucocorticoids (corticosterone and cortisol) and androgen hormones (estrone, estradiol, and dihydrotestosterone).[5][6][7] A common mnemonic device to remember which part of the adrenal cortex secretes which type of hormone is "salt-sugar-sex," referreing to mineralocorticoid (salt regulation) production in the zona glomerulosa, glucocorticoid (sugar metabolism) production in the zona fasciculata, and androgen (sex hormone) production in the zona reticularis.[8]
Development
During fetal development, aldosterone plays a role in maternal volume expansion necessary to accommodate fetal perfusion and may also increase the expression of placental growth factors.[9]
Congenital issues of concern related to aldosterone synthesis include autosomal recessive deficiencies in the enzymes responsible for adrenal hormone production. Congenital adrenal hyperplasia (CAH) can take many forms, depending on which enzyme is deficient. The three main enzymes that affect aldosterone are 21-hydroxylase, 11-beta-hydroxylase, and aldosterone synthase. A deficiency of any of these enzymes will halt aldosterone production. The production of aldosterone occurs in an interconnected pathway that produces mineralocorticoids, glucocorticoids, and androgens. The inability for aldosterone production to proceed leads to a buildup of intermediary products and cholesterol to be funneled down the glucocorticoid and androgen hormone production pathways instead. Depending on the severity of the enzyme deficiency, this can result in hyponatremia, hyperkalemia (due to the inability to exchange sodium for potassium in the nephron), and hypovolemia (low sodium causes a decrease in extracellular fluid). In aldosterone synthase deficiency, many of the functional losses are mitigated by the continued production of corticosterone, which acts similarly to aldosterone. The shunting of cholesterol toward the 17-alpha-hydroxylase pathway (androgen hormone production pathway) can result in virilization and ambiguous genitalia in females. Conversely, in the case of 17-alpha-hydroxylase deficiency, cholesterol is shunted towards mineralocorticoid production while glucocorticoid and androgen production is impaired causing ambiguous genitalia in genetic males and lack of secondary sexual development in genetic females. [10][8]
Organ Systems Involved
The organs involved in the production, utilization, and regulation of aldosterone are the adrenal glands, kidneys, and lungs due to the pulmonary conversion of angiotensin 1 to angiotensin 2 in the renin-angiotensin-aldosterone system.[4]
Function
When a stimulus such as low blood pressure or low serum sodium triggers the RAAS, first renin is secreted from the renal juxtaglomerular cells, then angiotensinogen is cleaved into angiotensin I. Angiotensin-converting enzyme (ACE) from the lungs then converts angiotensin I to angiotensin II, which in turn stimulates the production of aldosterone.[11]
Aldosterone is a mineralocorticoid steroid hormone that modulates activity directly and indirectly in the aldosterone-sensitive distal nephron which includes the late distal convoluted tubule, the connecting tubule, and the collecting duct system.[1][12] Because this region is so distal, aldosterone affects the final stages of electrolyte and water absorption within the nephron before the tubule contents are excreted in the urine. This only accounts for about 5-10% of total sodium reabsorption.[13]
Depending on the specific physiologic parameters, aldosterone may:
- increase sodium reabsorption
- increase water retention
- increase potassium excretion
- increase acid (H+) excretion
- increase bicarbonate (HCO3-) excretion and chloride reabsorption[1][12]
Although predominantly known for its activity in the kidney, aldosterone may act at mineralocorticoid receptors in other tissues as well such as the gastrointestinal tract, respiratory epithelium, myocardium, and vascular smooth muscle.[14]
Mechanism
As with all steroid hormones, aldosterone passes through cell membranes to bind to cytoplasmic receptors which translocate to the nucleus to influence mRNA transcription and subsequently protein synthesis. The mineralocorticoid receptor (MR) may have higher or lower affinity for for aldosterone depending on whether or not it is phosphorylated. In the phosphorylated state, MR has lower affinity for aldosterone therefore phosphorylation of MR in a given cell inhibits aldosterone activity.[1]
Within the principal cells of the late distal tubule and collecting ducts, MR is largely in a non-phosphorylated state. In principal cells, aldosterone increases the expression of sodium channels and sodium-potassium ATPase in the cell membrane. The sodium channels are on the luminal side of the principal cells and allow sodium to passively diffuse into the principal cells due to the transepithelial potential difference of -50 mV. This gradient is maintained by the sodium-potassium ATPase on the basolateral side, which uses ATP to actively transport sodium into the blood and potassium into the cell. Meanwhile, potassium channels on the luminal side of the cell that allow passive diffusion out of the cell into the lumen of the kidney whenever a sodium ion enters the cell. The net effect of this process is sodium absorption from the lumen, which allows for water absorption, assuming ADH is present to make the cells permeable to water. This directly results in an increase in osmolality within the blood, causing water to flow down its concentration gradient.[15]
Within intercalated cells, MR is often in a phosphorylated state therefore the dephosphorylation of MR in the presence of angiotensin II enables intercalated cells to be responsive to aldosterone. This conditional response of intercalated cells results in seemingly paradoxical effects of aldosterone dependent upon whether or not angiotensin II is present.
In alpha-intercalated cells (A-intercalated, acid-secretory), aldosterone increases the expression of apical hydrogen ATPases to stimulate hydrogen ion (proton) excretion into the lumen. Additionally, the sodium resorption from adjacent principal cells creates a more negatively charged lumen space which further encourages acid secretion from the intercalated cells to compensate.
Within the non-A intercalated cells (beta-intercalated, and non-A non-B intercalated cells), aldosterone increases the activity of apical bicarbonate-chloride exchangers, which reabsorb chloride from the lumen into the cell and excrete bicarbonate from the cell into the lumen.[1][12]
Related Testing
The most common test to assess disturbances of the aldosterone pathway is the aldosterone: renin ratio. This determines whether there is an isolated aldosterone problem or there is a disturbance within renin-angiotensin system. If an aldosterone problem is suspected, and the results show no elevation in either aldosterone or renin, then congenital adrenal hyperplasia is suspected. If both aldosterone and renin are increased, and their ratio is less than 10, then the differential includes renovascular hypertension. If the renin value is normal, the aldosterone level is elevated, and the ratio is greater than 30, the differential includes Conn syndrome. This can be confirmed with a salt suppression test, an MRI of the adrenal glands, and adrenal vein sampling.
Pathophysiology
The release of aldosterone from the adrenal glands is regulated via the renin-angiotensin II-aldosterone system. This system is initially activated via a decrease in the mean arterial blood pressure to increase the blood pressure. The decrease in blood pressure is initially sensed within the afferent arterioles of the kidney. Prorenin is then released by mechanoreceptors and is converted to renin by the juxtaglomerular cells (JG cells). The JG cells can also release renin after sympathetic stimulation of their beta one receptor. Renin is the enzyme that converts angiotensinogen to angiotensin I. Angiotensin I then is converted to angiotensin II in the lungs and kidneys by angiotensin-converting enzymes (ACE). Angiotensin II is an octapeptide that is activated by type-1, G protein-coupled angiotensin II receptors. These receptors have different functions depending on the types of cells that contain the receptor. However, it has five primary functions that include: increasing aldosterone, increasing sodium-hydrogen exchange within the proximal renal tubule, increasing thirst drive within the hypothalamus, increasing antidiuretic hormone, and acting on G protein-coupled receptors that activate IP3/Ca2+ second messenger systems within arterioles to cause vasoconstriction. Aldosterone then undergoes its actions within the kidney.
Clinical Significance
Aldosterone is clinically significant for two reasons. An increase or decrease in aldosterone can cause disease and medications affecting its function alter blood pressure. Changes in the concentration of aldosterone, either too much (Conn syndrome and renovascular hypertension) or too little (certain types of Addison's disease and congenital adrenal hyperplasia), can result in disastrous effects on the body.[16][15]
Hyperaldosteronism is caused by either a primary tumor within the adrenal gland (Conn syndrome) or via renovascular hypertension. A primary tumor within the adrenal gland causes an uncontrolled production and release of aldosterone. Renovascular hypertension increases aldosterone through two primary mechanisms: fibromuscular dysplasia (usually in young females) and atherosclerosis (usually in older individuals). Both decreased perfusion to the afferent arterioles of the kidney causes the renin-angiotensin system to be activated. This causes uncontrolled hypertension and hypokalemia.
Addison’s disease is characterized by a hypo-functioning adrenal gland. However, depending on the cause of Addison’s disease, the regulation of aldosterone may be unaffected. Aldosterone is only affected by Addison’s disease when the adrenal gland undergoes destruction, for example, in autoimmune-mediated destruction. Aldosterone is controlled by the renin-angiotensin system, while the rest of the adrenal glands' hormone production is controlled by adrenocorticotropic hormone (ACTH). Therefore, in cases of Addison’s disease caused by pituitary dysfunction, adrenal insufficiency will exist, but with appropriate aldosterone levels. This is due to the fact the renin-angiotensin system remains intact.
Contraction alkalosis is a side effect of increased absorption of water via aldosterone and ADH pathways during a volume-depleted state. The body senses a low mean arterial blood pressure when the ECF is low. Therefore the renin-angiotensin system is activated. This causes an increase in water absorption as well as activation of aldosterone. Aldosterone causes sodium to be absorbed and potassium to be excreted into the lumen by principal cells. In alpha intercalated cells, located in the late distal tubule and collecting duct, hydrogen ions and potassium ions are exchanged. Hydrogen is excreted into the lumen, and the potassium is absorbed. This mechanism prevents the body from losing too much potassium, which causes a relative depletion of hydrogen ions in the blood causing an alkalotic state.
Medications
Beta-agonists/antagonists can cause the increase or decrease of renin by the JG cells, which translates to an increase or decrease in aldosterone.
Angiotensin-converting enzyme inhibitors, a common class of hypertensive medications, blocks ACE from producing angiotensin II which results in a decreased aldosterone.
Angiotensin II receptor blockers (ARBs) block angiotensin II receptors which result in a decrease in aldosterone. This class of medications is used to control blood pressure when a patient has an intolerance to ACE-I.
Spironolactone is an aldosterone receptor blocker, which prevents aldosterone from acting on the receptors within the principal cells of the kidney.
Amiloride and triamterene are medications that block the sodium channels on the luminal side of the principal cells within the kidney. This prevents aldosterone from exerting its full effect on the kidney.
Generally speaking, all of these medications block the function of aldosterone, which prevents sodium absorption and potassium excretion. Therefore, possible side effects to all of these medications are hyponatremia, hyperkalemia, and hypovolemia.