Continuing Education Activity
Menkes disease is a rare X-linked recessive progressive multisystemic disease of copper metabolism. Patients usually exhibit a severe clinical course with death in early childhood. Early diagnosis of Menkes disease is clinically very challenging because of the subtle clinical features and nonspecific biochemical markers. Accurate diagnosis is essential both for proper management to reduce morbidity and mortality and also for parental counseling and prenatal diagnosis. This activity reviews the pathophysiology, clinical features, evaluation, and treatment of Menkes disease and highlights the role of the interprofessional team in the management.
Objectives:
Describe the etiology of Menkes disease.
Outline the typical clinical features and explain the evolution of a patient with Menkes disease.
Review the management options available for Menkes disease.
Summarize the importance of collaboration and coordination among the interprofessional team to enhance patient care and improve outcomes in Menkes disease.
Introduction
Menkes disease is a rare X-linked recessive progressive multisystemic disease of copper metabolism. Patients usually exhibit a severe clinical course with death in early childhood. Early diagnosis of Menkes disease is clinically very challenging because of the subtle clinical features and nonspecific biochemical markers. Accurate diagnosis is essential for proper management to reduce morbidity and mortality and also for parental counseling and prenatal diagnosis.[1]
Menkes et al first described the disease in 1962.[2] In 1972, Danks et al first observed that copper metabolism was abnormal. In 1973, they noted the similarity between kinky, wiry, or steely hair and the brittle wool of sheep found in copper-deficient areas of Australia. Later, they reported abnormal copper and ceruloplasmin levels in these patients.[3][4]
Etiology
In Menkes disease, the underlying abnormality in copper metabolism is secondary to a mutation in the ATP7A gene located on Xq13.3, which has 23 exons. This gene encodes 1500 amino acids and is abundantly expressed in various organs, such as the brain, lungs, kidneys, and muscles.[5] So far, about 357 different mutations, including insertions, deletions, partial deletion, missense mutations, and splice mutations, have been identified in the ATP7A gene.[6] Genetic studies indicate that mothers of 75% of patients are carriers, and the remaining 25% are not. There is no apparent correlation between mutations and the clinical course. As expected, most patients are male, but few female patients have been reported. It is likely due to X autosome translocation, where normal X is preferentially inactivated. Other causes are point mutation and skewed inactivation of the normal X chromosome.[7]
Epidemiology
The incidence of Menkes disease is close to 1 in 35,000 live male births.[6] In the United States, the incidence ranges between 1 in 50,000 and 1 in 250,000; one-third of these cases result from new mutations. Between 1993 and 2003, a study conducted in Japan found that the incidence of Menkes disease was 1 in 2.8 million live births. The incidence in male live births was noted to be 4.9 per million.[8]Females are usually carriers, but cases have been reported due to unusual genetic circumstances. The incidence in Australia is much higher (1 in 50,000 to 1 in 1,00,000), possibly due to the founder effect.[7]
There is no racial or ethnic predilection found for Menkes disease. The genetic theory proposes that one-third of cases with Menkes disease exhibit new mutations. These de novo mutations can occur anywhere, independent of race or ethnicity.
Menkes disease occurs in males almost exclusively due to the X-linked recessive trait. Females are carriers and usually do not manifest symptoms unless there are unusual genetic circumstances.
Patients with Menkes disease present between six to eight weeks after birth when parents notice delayed development or the look of an unusual eye or unusual limb movements indicative of seizure activity.
Pathophysiology
ATP7A gene encodes ATP7A, which is an active copper (Cu) transporter. ATP7A is a transmembrane protein predominantly expressed in enterocytes, the placenta, and the central nervous system, where, at a steady state, it directs Cu to Cu-dependent enzymes. In low to normal copper states, the ATPase is localized in the trans-Golgi network (TGN), which is retained by Acetyl-CoA transporter 1. In the TGN, ATP7A transfers copper from the cytoplasm into the Golgi complex for the metalation of multiple cuproenzymes. If copper levels are high, it effluxes out of the cell by relocating to the plasma membrane.[9]
The clinical features of Menkes disease are either a direct consequence of enzyme dysfunction or secondary to an inability to load these enzymes with Cu.[10] The failure of ATP7A in the enterocytes leads to subsequent failure of the efflux pump and causes excessive Cu accumulation in the enterocytes with generalized Cu deficiency. The enzymes requiring copper for important biochemical functions include cytochrome C oxidase, dopamine beta-hydroxylase, lysyl oxidase, tyrosinase, superoxide dismutase, and peptidyl glycine alpha amid monooxygenase.[11][12] The cytochrome oxidase deficiency affects cellular respiration and causes CNS degeneration and ataxia. Autonomic symptoms, including hypotension and hypothermia, are due to abnormal catecholamine synthesis from deficient dopamine beta-hydroxylase. Menkes disease also exhibits marked connective tissue abnormalities, including skin, joints, and bone abnormalities, and impaired blood vessel integrity due to the absence of lysyl oxidase and defective cross-linking of collagen and elastin.[13] Skin hypopigmentation and hair abnormalities are due to abnormal tyrosinase and sulfhydryl oxidase, respectively.
Profound neurological features are due to low Cu in the brain. Likely explanations include:
- Cu gets trapped in the blood-brain and blood-cerebrospinal fluid barriers, while glial cells and neurons are deprived of Cu.
- Generally, Cu is the noncompetitive antagonist of NMDA receptors. Normally, ATP7A mediates the availability of NMDA receptors and a releasable pool of copper in hippocampal neurons. The absence of ATP7A markedly accentuates the NMDA receptor excitability in the neurons, causing seizures.[14]
- The deficiency of cytochrome C-oxidase causes accumulations of superoxide radicals and neuronal damage.
- Impaired blood vessel integrity with secondary ischemia leading to infarction due to deficiency of lysyl oxidase.
Histopathology
Histopathological findings in both hair and brain have been described in Menkes disease. Findings of hair on light microscopy include pili torti (180 degrees twisted on-axis), monilethrix, and trichorrhexis nodosa. The internal elastic lamina is fragmented with the proliferation of the intimal layer.[13]
Neuropathological changes in the brain include striking cortical neuronal loss, mineralized neurons, gliosis, subcortical myelin loss associated with severe axonal degeneration, and widespread atrophy of grey and white matter present. Light microscopy findings include focal degeneration that extends to all layers of the cerebral cortex with more prominent neuronal loss in the hippocampus, striatum, hypothalamus, and thalamus. Granular neurons are severely depleted in the cerebellum with relative preservations of Purkinje cells, which differentiate this from other neurodegenerative conditions. Purkinje cells branch in the molecular pattern, forming cactus-shaped extensions. The neuropathological changes are largely due to the deficiency of cytochrome C oxidase.[15][16]
Light microscopy findings in patients with hepatomegaly show nonspecific cholestasis and focal necrosis. Light and electron microscopy findings of ocular tissue show progressive degeneration of retinal ganglion cells, loss of nerve fibers, and optic atrophy.
History and Physical
Classical Menkes disease patients usually exhibit a severe clinical course with death in early childhood, typically by age 3. Progressive neurodegeneration and connective tissue dysfunction characterize the clinical picture of classical MD.
Pregnancy and labor are usually uncomplicated, with normal anthropometry at birth. Clinical features in the newborn period include prolonged jaundice, hypothermia, hypotonia, and feeding difficulties at birth. Spontaneous fractures and cephalohematoma may present at birth.[1][17][18] The typical symptoms usually start by two months with characteristic skin and hair findings. Hair is normal at birth but is replaced by fine, sparse, kinky, wiry, or steely hair. Sagging facial features, micrognathia, cutis laxa, and blue iris are noted. Various congenital malformations have been reported, including congenital microblepharia, entropion, long arched palate, cerebellar hypoplasia, cystic changes in the lungs, and complete AV block.[19]
Neonatal Menkes disease is often missed because of the more nonspecific clinical and biochemical findings. The patient may present with hypoglycemia, hypothermia, prolonged jaundice, cephalohematoma, and pectus excavatum at birth. Characteristic hair findings are often absent, and microscopic findings of hair are not evident in the early phase of life. A definite diagnostic test, copper egress in cultured fibroblast, takes several weeks to result, so a more rapid and reliable test that diagnoses the patient before the onset of symptoms is critical. Such newly recognized tests include plasma catecholamine analysis and placental copper concentrations.
Neurological manifestations are the most common and characteristic of Menkes disease. Developmental regression and seizures are usually the first to start around 2 to 3 months.[20] Infection or febrile state triggers the onset of seizures. The disease can present in other ways, like nonconvulsive seizures and recurrent episodes of apnea. The evolution and progression of seizures in Menkes disease follow three different stages. The early stage usually manifests at 2 to 3 months as focal clonic seizures leading to status epilepticus, usually in the setting of fever. Fluctuating disturbance in the blood supply related to the tortuosity of vessels may be the cause. The intermediate stage typically starts with a mean age of 9.5 months as an intractable infantile spasm. A likely explanation would be neuronal degeneration in the subcortical structures, such as basal ganglia and thalamus. Late-stage presents at a mean age of 24 months with multifocal myoclonic seizures and tonic spasms due to progressive cortical degeneration.[21] Other hallmark findings include neuroregression. The patient develops motor skills to some extent, with no language skills. There is also an association between growth restriction and failure to thrive.
Connective tissue dysfunction and skeletal abnormalities are very common in Menkes disease. Tortuous blood vessels and fluctuating blood supply are some of the well-established causes of seizures. Internal jugular vein dilatation or phlebectasia and aneurysm of brachial arteries have been reported.[22] The defective connective tissue formation also manifests as loose, wrinkled skin. Motor development is also delayed secondary to muscular hypotonia and other connective tissue disorders. Bone deformities include wormian bones in the skull, pectus excavatum or pectus carinatum, osteopenia, and long bone fractures. Metaphyseal fractures, rib, and skull fractures in these patients are often misdiagnosed as child abuse. Pamidronate treatment is effective in increasing bone mineral density and managing osteoporosis.[23]
Ocular findings include aberrant eyelashes, very poor visual acuity, strabismus, blue irides, iris stromal hypoplasia, and peripheral retinal hypopigmentation.[24]
Menkes disease patients present with dysautonomia symptoms (chronic diarrhea, fainting, orthostatic hypotension) attributed to abnormal catecholamine synthesis due to defective dopamine beta-hydroxylase. Gastrointestinal manifestations are wide and may cause gastric polyps, reflux disease, colonic diverticula, and progressive hiatal hernia. Reports of hepatomegaly in Menkes disease have been made. Patients with significant growth failure and GI complications often need a gastrostomy tube.
Urological complications are very frequent in Menkes disease, and the most common manifestation is bladder diverticula. Bladder diverticula often leads to frequent urinary tract infections, ascending infections, hydronephrosis, and renal parenchymal damage. Cases of renal rupture, vesicoureteric reflux, and cryptorchidism have been reported.[18] Recurrent infections and pneumonia are reported to be some of the common causes of morbidity and mortality in Menkes disease. It can be due to defective free radical clearance, diffuse pan-lobular emphysema, and cystic changes with abnormal pulmonary vascular development.
Evaluation
The diagnosis of Menkes disease is made by the characteristic clinical, biochemical, and radiological features. The characteristic clinical features often suggest the diagnosis.
Biochemical evaluation plays a significant role in the diagnosis of Menkes disease. Serum copper and ceruloplasmin levels are typically low in Menkes disease in the range of 0-55 microgram/dl and 10-160 mg/dl, respectively, with the normal levels being 70 to 150 micrograms/dl and 200-450 mg/dl. These levels are typically low in babies less than six months. So, biochemical labs should never be interpreted alone without other testing. In this scenario, plasma catecholamine analysis indicative of dopamine beta-hydroxylase deficiency can be used as a rapid diagnostic test.
Features of hair on light microscopy are often very characteristic in the case of Menkes disease. Examining as many strands as possible is essential because every hair may not demonstrate typical morphological features.
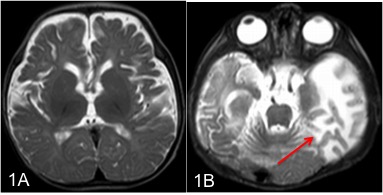
Neuroimaging has a major role in establishing the diagnosis of Menkes disease. Cerebral atrophy and delayed myelination are the most commonly observed features on the MRI brain. (Fig.1A) Other features include subdural effusion, hematoma and hygroma, leukoencephalopathy, basal ganglia signal changes, and intracranial vessel tortuosity.[25][26] Focal tumefactive white matter lesions on MRI are especially seen before ten months of age. (Fig.1B) Hyperintense drop-shaped lesions in centrum semiovale, with restricted diffusion, also have been reported.[27] Cerebral infarctions can occur due to thrombotic occlusion and artery-to-artery embolism due to erratic and turbulent blood flow in a tortuous blood vessel. In addition to the above findings, extensive arterial tortuosity is highly suggestive of Menkes. (Fig.2A&B) These vascular changes are more prominent in MRA than in routine MRI.[28][29]
EEG in Menkes follows three characteristic stages with the progression of the disease. Ictal findings in the early stage include slow spike waves and slowing in the posterior region, whereas interictal EEG shows multifocal polymorphic waves. The intermediate stage shows multifocal hypsarrhythmia with diffuse irregular slow and spike waves. The late stage is characterized by multifocal high-amplitude activity and mixed irregular slow waves.[30]
X-ray findings include generalized osteoporosis, metaphyseal flaring, and spurs in long bones. Periosteal diaphyseal reactions and thickenings are present. Rib fractures are often misdiagnosed as child abuse. Menkes disease should be considered as a differential diagnosis in infants with unexplained subdural hematoma in the absence of retinal hemorrhage.[31]
Molecular genetic testing includes single-gene testing and multigene panels. The sequential analysis includes analyzing ATP7A followed by gene-targeted deletion/duplication analysis if no pathogenic variant is found. The clinician can customize multigene panels, including ATP7A and other genes of interest, such as FBLN5-related cutis laxa and ELN-related cutis laxa while limiting the identification of variants of uncertain significance and variants that do not explain the underlying phenotype.[32]
Prenatal diagnosis is available for women with carrier states, including chorionic villi sampling and analyzing copper intake in the cultured fibroblast, especially if the genetic mutation is unknown. If an affected patient in the family has a known mutation, then specific genes can be tested.
Treatment / Management
Menkes disease is a fatal disease with death usually occurring between 6 months to 3 years. Treatment outcome depends on the severity of ATP7A gene deletion and residual functioning ATP7A protein. When designing a treatment plan for patients with Menkes disease, providers should take into account three basic principles:
- Bypass the block in enteral absorption of copper
- Copper should be made available to the cellular enzymes that need it as a cofactor
- To avoid irreparable neurodegeneration in Menkes disease, infants should be identified, and treatment must be commenced earlier in life.
The goal of the treatment is to provide copper to the copper-dependent enzymes. Early treatment of Menkes disease is essential (ideally within four weeks of life). The copper histidine component has been reported to be the most effective. Oral copper treatment is not effective because it gets trapped in the enterocytes. The subcutaneous or intravenous route is preferred. Neurodegeneration can be effectively reduced in some patients if the treatment is started at less than three weeks of age. Copper-histidine treatment can effectively reverse skin and hair changes, improving appendicular tone, weight gain, and achieving socio-cognitive milestones. Outcomes also depend on the initiation of treatment and residual functioning ATP7A.[33][34] Significant secondary mitochondrial dysfunction is also noted to be common in untreated individuals and subsequently normalized, followed by corrections of serum copper in response to replacement therapy.
Menkes disease needs multiple specialists in the management of complications. For example, a feeding tube may be given to ensure proper nutrition, a G tube for a patient with significant dysphagia, surgical correction for urological complications, and seizure management with multiple antiepileptics. In addition to medical and surgical management, the patient needs extensive physical, occupational, and speech therapies. Orthopedic aids may improve symptoms due to connective tissue problems.
Although replacing copper does not ensure substantial neurologic improvement in all cases of Menkes disease, its use leads to modest clinical benefits, such as reduced seizure frequency and decreased irritability.[35] The decision-making process should mainly include a frank discussion with the parents, as there are very limited benefits to be expected from copper therapy. Copper replacement is definitely indicated in cases where the diagnosis is established before the neurologic damage begins to occur, as the prevention of further neurodegeneration is possible in some individuals.
Damage to the proximal renal tubule is a known complication of copper overload.[36] This effect could be attributed to the natural tendency of Menkes disease patients to sequester copper in their kidneys. However, this adverse effect is not clinically significant in most cases as renal losses barely get to a point where a replacement, such as oral bicarbonate, becomes indicated.
Other treatment options that have been studied but still need more evidence to become established management options include the following:
- Vitamin C may help limit copper and metallothionein interaction[37]
- Vitamin E
- Carbamic acid derivative - diethyldithiocarbamate
- Brain-directed viral gene therapy[38]
In Menkes disease, there is no heightened anesthetic risk.[39][40] The following surgical situations may arise in Menkes disease:
- Myringotomy tubes - for chronic otitis media
- Gastrostomy tube - for feeding problems
- Bladder diverticula repair
Differential Diagnosis
Occipital horn syndrome (OHS): OHS is the milder form of Menkes disease that usually manifests at 5 to 10 years of age with overlapping features of classical Menkes. At birth, infants present with hypothermia and prolonged jaundice. Connective tissue abnormalities are prominent in OHS, including umbilical and inguinal hernias. A characteristic feature of OHS is bony abnormalities of bones, typically symmetric exostoses from occiput. Neurological symptoms are less profound than Menkes, intellectual capabilities are usually low to borderline, and motor delays are secondary to hypotonia.[41]
ATP7A-related distal motor neuropathy: Adult-onset distal motor neuropathy similar to Charcot Marie tooth disease. It is characterized by progressive distal motor neuropathy with minimal to no sensory loss. Distal muscle weakness and atrophy get more prominent and may occasionally cause pes cavus. Biochemically, the levels are normal.[34]
Biotinidase deficiency: Typically occurs in the first few months of age. Presents with seizures, hypotonia, global developmental delay, ataxia, vision, and hearing problems. Skin rashes, hypopigmentation, and hair loss are common.
Hupke-Brendel syndrome: Rare autosomal recessive conditions characterized by bilateral congenital cataracts, sensorineural hearing loss, and severe developmental delay. Patients usually die between 9 months and 6 years of age. Diagnosis is usually established in a proband with characteristic features and very low serum copper and ceruloplasmin levels and biallelic pathogenic variants in SCL33A1 are identified by molecular genetic testing.[42]
Prognosis
The life span of patients with Menkes disease is difficult to predict, although the majority of these children do not live past the age of three years. A common cause of death is pneumonia, which leads to respiratory failure. However, some patients die suddenly without any apparent acute medical cause. Major underlying causes of morbidity and mortality in Menkes disease are associated with the following systems:
- Neurologic
- Gastrointestinal
- Connective tissue
- Vascular system
Complications
Late complications of the disease include intractable seizures, subdural hematoma, blindness, and recurrent infections. In classical Menkes disease, death occurs by three years of age, usually secondary to vascular complications or respiratory infections. Renal complications may also occur secondary to copper sequestration in the proximal renal tubule. Growth failure becomes apparent shortly after the beginning of neurodegeneration.
Ocular pathologies have been observed, such as retinal hypopigmentation, partial optic nerve atrophy, macular dystrophy, congenital cataracts, and microcysts in the iris.
Consultations
To provide the best management plan to patients with Menkes disease, an interprofessional approach is critical, preferably at a common location, such as a pediatric center. The following specialties should particularly be consulted:
-
Medical geneticist
-
Neurologist
-
Gastroenterologist and nutritionist
-
Urologist
-
Otolaryngologist
-
Dentist [43]
-
Psychologist/social worker
-
Physical and occupational therapist
Deterrence and Patient Education
Genetic counseling and prenatal diagnosis can be very helpful in preventing Menkes disease. However, in one-third of all cases of Menkes disease, new mutations are the underlying pathology. There are established guidelines for prenatal screening and diagnosis.[44]
Physical and occupational therapy techniques may be taught to parents. Menkes disease impacts are severe on the families, and psychosocial support is often valuable and crucial. Families of patients with Menkes have to endure the pain of transitioning from apparent good health to irrevocable debilitating illness within the first few months of life. Therefore, psychological support is an essential part of a holistic management plan.
Enhancing Healthcare Team Outcomes
Menkes disease has no specific curative treatment, and it is a progressive multisystem disease. Also, currently, there are no newborn screening programs available. Early diagnosis of Menkes disease is clinically very challenging because of the subtle clinical features and nonspecific biochemical markers. So, a high index of suspicion is required when evaluating a newborn with hypotonia, hypothermia, and jaundice, especially in the setting of significant family risk factors or connective tissue abnormalities, because early replacement of copper reverses and prevents complications.
Menkes disease is better managed by an interprofessional team approach, including a neonatologist, pediatric neurologist, geneticist, nutritionist, physiotherapist, and occupational therapist. Genetic counselors play a significant role in parental reproductive risk counseling and prenatal diagnosis. Menkes disease families need medical, educational, psychosocial, and financial support services in their local communities.