Introduction
In the human embryo, eyes develop through a complex and delicate process. Interruption of this process leads to malformations of the eye or related structures that can result in both functional and aesthetic deficits. Congenital ocular malformations are relatively rare, with approximately 5 in every 10,000 live births being affected. These conditions range from having little impact on the life and function of the child to being highly debilitating. Some of the more common malformations include anophthalmia, microphthalmia, coloboma, aniridia, optic nerve hypoplasia, congenital cataracts, corneal opacity, and congenital glaucoma.[1][2][3]
Development
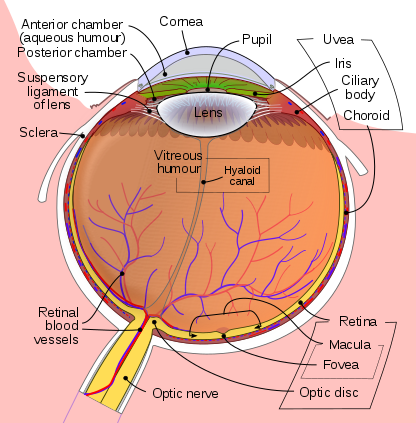
Eye development occurs in the human embryo from approximately the third week through the tenth week of gestation. Ocular tissues are of mesodermal and ectodermal origin. The retina, ciliary body, optic nerves, and iris are derived from neuroepithelium. The lens, eyelid, and corneal epithelium are formed from surface ectoderm. The sclera, blood vessels, ocular muscles, vitreous, and corneal endothelium and stroma, develop from the extracellular mesenchyme. The homeobox gene Pax6 is vital to the initiation of eye development, which begins with evagination of the optic grooves. During the fourth week of pregnancy, upon neural tube closure, the optic grooves are transformed into optic vesicles, which are outgrowings of the brain on either side of the forebrain. The optic vesicles make contact with the surface ectoderm which induces changes in the ectoderm necessary for further development. The optic vesicles develop into the optic cup, the inner layer of which forms the retinal pigment epithelium, with the retina being formed by the outer layer. The iris and ciliary body develop from the middle part of the optic cup. As the optic cup invaginates, the lens placode forms through the thickening of the ectoderm and eventually forms the lens vesicle upon separation from the ectoderm.
The choroid fissure is a space at the inferior portion of the optic vesicle that accommodates the passage of blood vessels to the eye. Wingless-related integration site (Wnt) and fibroblast growth factor (FGF) are important protein regulators of neuroectoderm development during early morphogenesis. Bone morphogenic proteins (BMP) are heavily involved in the regulation of optic cup development. Regulation of ocular embryogenesis is vital to proper development; Sonic hedgehog reduces Pax6 expression, and retinoic acid (a vitamin A derivative) plays an important role in paracrine signaling.[4][5][6]
Pathophysiology
Congenital ocular defects arise through a variety of mechanisms and in association with many different syndromes and diseases. The Pax6 gene has been identified as one of vital importance to the initiation of eye development and its proper progression. Many genetic mutations and environmental factors are implicated in ocular malformations.
Clinical Significance
Anophthalmia, or the absence of one or both eyes, is a rare result of genetic mutations, chromosomal abnormalities, or prenatal environmental factors. Anophthalmia presents as a small, bony orbit, malar prominence, reduced palpebral fissure, short eyelids, and a constricted mucosal socket. Sex-determining region Y-box 2 (Sox2) anophthalmia syndrome follows an autosomal dominant inheritance pattern and results from a mutation in the Sox2 gene which prevents the associated protein production. Retinol-binding protein 4 (RBP4) mutation also causes autosomal dominant anophthalmia, mediated through increased fetal susceptibility to vitamin A deficiency. Other associated genes include orthodenticle homeobox 2 (OTX2), CEH10 homeodomain-containing homolog (CHX10), retina and anterior neural fold homeobox (RAX), and bone morphogenic protein 4 (BMP4). Environmental factors that have been implicated in anophthalmia include gestational-acquired infections, thalidomide exposure, maternal vitamin A deficiency, gestational radiation exposure, and maternal solvent abuse. Deletions of q22.1 through q22.3 on chromosome 14 have also been shown to cause anophthalmia. Anophthalmia can be diagnosed prenatally using ultrasound and amniocentesis. Treatment options include cosmetic surgery and the use of a prosthetic eye.
Microphthalmia describes a condition in which one or both eyes are unusually small and have anatomical abnormalities. Microphthalmia has associations with environmental factors such as fetal alcohol syndrome, herpes simplex virus, cytomegalovirus, and rubella infections; genetic causes including chromosomal abnormalities such as trisomy 13, Wolf-Hirschhorn syndrome, 13q deletion, and triploid syndrome; and monogenetic mutations which may follow autosomal dominant, autosomal recessive, or X-linked inheritance patterns. While many genes have been implicated in microphthalmia presentation, Sox2 and microphthalmia-associated transcription factor (MITF) have the best-documented effect. MITF is located on chromosome 14, and its failure of expression prevents full differentiation of the pigmented retina, causing a malformation of the choroid fissure and drainage of the vitreous fluid, which inhibits ocular enlargement.
Coloboma refers to a hole in an ocular structure, such as the iris, choroid, retina, or optic disc, that is present from birth. Failure of the choroid fissure to fuse causes a coloboma. The severity of functional impairment varies widely from not affecting vision to causing complete blindness. A coloboma can occur unilaterally or bilaterally; most commonly, only the iris is affected. In some cases, colobomas are associated with microphthalmia, glaucoma, nystagmus, scotoma, and/or strabismus. Identified causes of colobomas include a mutation in the paired box gene 2 (PAX2) gene and fetal alcohol syndrome. Treatment varies by case, but some options include surgical repair of an iris defect, placement of a prosthetic iris device, use of a specialized cosmetic contact lens, and refractive correction with contact lenses, glasses, or laser surgery.
The complete absence of the iris is described as aniridia, which usually presents with bilateral involvement. Aniridia can be associated with hypoplasia of the macula and optic nerve, cataracts, corneal changes, nystagmus, amblyopia, and buphthalmos. It can severely impair vision and can present as a component of syndromes such as WAGR syndrome (Wilms tumor-aniridia syndrome), and Gillespie syndrome. As stated previously PAX6 is important for the proper development of the eye, and its mutation may lead to aniridia. Both hereditary and sporadic forms of aniridia exist, with most hereditary forms following an autosomal dominant inheritance pattern. Cases of autosomal recessive aniridia rarely occur, an example of which is Gillespie syndrome. WAGR syndrome can result from sporadic mutation of the Wilms Tumor 1 (WT1) gene.
Optic nerve hypoplasia (ONH) presents as an abnormally small optic disc due to the improper development of optic nerve axons. It is commonly associated with endocrinopathies, other developmental abnormalities, and other brain malformations. ONH is diagnosed through fundoscopic examination, though it is difficult to make an accurate prediction of visual acuity based on an observation of optic nerve size, as visual acuity is more closely related to the macular fiber integrity. Small disc diameter is defined as having a DD/DM ratio of less than 0.35. Strabismus and nystagmus are associated with various presentations of ONH. While there are not documented specific genetic mutation causes of ONH, mutations of SOX2 and homeobox expressed in embryonic stem cells 1 (HESX1) have a possible association. Hypopituitarism due to hypothalamic dysfunction occurs in approximately 75% of ONH cases. Other endocrine abnormalities include elevated serum prolactin, growth hormone deficiency, hypothyroidism, adrenocorticotropic hormone deficiency, and diabetes insipidus. MRI and endocrinologic and pituitary function tests are conducted in suspected cases of ONH.
Congenital cataracts, lens opacities present at birth, occur in a wide variety of disorders. Morphologic configurations of congenital cataracts include polar, sutural, lamellar, coronary, cerulean, nuclear, membranous, capsular, and complete. Some congenital cataracts are associated with other features of more extensive diseases, while some are isolated inherited traits. There are many causes of congenital cataracts including genetic and metabolic etiologies, infections, teratogen exposure, and developmental anomalies. Some of the anomalies associated with congenital cataract formation include aniridia, anterior segment dysgenesis, persistent fetal vasculature, and posterior lenticonus. Significant cataracts are treated with surgical removal of the opacified lens.
Congenital corneal opacity can occur through a variety of processes. It is associated with corneal dystrophies, anterior segment dysgenesis, infections, corneal dermoids, congenital glaucoma, delivery trauma, metabolic disorders, posterior corneal defects, microphthalmia, and some developmental syndromes such as Axenfeld-Rieger syndrome. Corneal opacities are typically identified by morphology as exhibiting central opacity, peripheral opacity, diffuse opacity, bilateral or unilateral clouding. Treatment generally involves surgery, with penetrating keratoplasty being among the most common corrective techniques. Management focuses on the treatment of concurrent issues as well as the prevention of amblyopia and long-term visual impairment.
Pathologically elevated intraocular pressure with onset during the first year of life is diagnosed as congenital glaucoma. It causes blindness in approximately 10% of cases. Congenital glaucoma is commonly accompanied by epiphora, blepharospasm, and photophobia. Edema of the corneal stroma can cause Haab striae in the Descemet membrane which can lead to corneal opacification. The etiology of primary congenital glaucoma is unknown, but risk factors include male gender, consanguinity, and affected siblings. Most cases are sporadic, but there are also autosomal recessive forms. Management focuses on the lowering of intraocular pressure with goniotomy or trabeculectomy being the most commonly employed surgical techniques. In refractory cases, trabeculectomy with mitomycin C or glaucoma implant surgery are options to be considered. Cycloablation may be used in resistant cases. Medical therapy can be useful in lowering pressure before surgery and in augmenting surgical effects when needed. Common medical therapies include carbonic anhydrase inhibitors, beta-blockers, prostaglandin analogs, and combination drugs.[7][8][9][10][11][12][13][14][15][16][17][18][19][20][21][22][23][24][25]