Continuing Education Activity
Renal cell carcinoma (RCC) is the most common malignant tumor of the kidney and constitutes over 90 percent of all renal malignancies. RCC may be sporadic (~96%) or familial (4%) and is a heterogeneous group of disorders that are sub-classified into several distinct sub-types associated with distinct genetic abnormalities. This activity reviews the evaluation and management of renal cell carcinoma and highlights the role of the interprofessional team in improving care for patients of renal cell carcinoma.
Objectives:
- Review the imaging findings associated with management considerations for patients with renal cell carcinoma.
- Describe the patient physical exam findings associated with advanced or metastatic renal cell carcinoma.
- Explain the staging system and its application to management options available for renal cell carcinoma.
- Outline the importance of collaboration and communication amongst the interprofessional team to ensure the appropriate follow-up during initial management and follow-up to enhance postoperative management and care.
Introduction
The most common type of cancer arising in the kidney is renal cell carcinoma (aka hypernephroma or Grawitz tumor), making up more than 9 out of 10 renal cancers in adults. Other types include transitional cell carcinomas of the renal pelvis, which behave like bladder cancers. Renal sarcoma is another rare tumor of the kidney. This review will focus mainly on renal cell carcinoma (RCC), which accounts for over 3% of all adult malignancies and has several histological subtypes. It is a tumor of older age group and is most commonly seen between the ages of 50 to 70 years that has an approximate 2:1 male to female ratio.[1]
Etiology
The strongest risk factor for renal cell carcinoma (RCC) is smoking and not just cigarette smokers but also pipe, and cigar smokers are at higher risk. Obesity (especially in women) is another major risk factor. It is thought that the incidence of kidney cancers can be halved by eliminating tobacco smoking and excess body weight. Several other factors also contribute to increased risk and include high blood pressure, chronic renal failure, and occupational exposure to certain chemicals, such as trichloroethylene. Moderate amounts of alcohol consumption (up to about two drinks per day), fruits and vegetables rich diet, and long term fatty fish consumption is associated with a reduced risk of kidney cancer.[1][2][3]
Current studies implicate the VHL gene in the development of both sporadic and familial type of clear cell-RCC (CCRCC).[4][5] Mutation in the MET gene is a feature of the familial type of papillary-RCC (PRCC).[4] Although sporadic PRCC has a much wider set of molecular alterations associated with it.[4][6][7]
About 4% of renal cell cancers are the result of rare hereditary conditions as detailed below:
- Von Hippel-Lindau (VHL) Syndrome: is associated with the development of the formation of cysts and tumors in various organs e.g., pancreatic neuroendocrine tumors, pancreatic cysts, cerebellar and spinal hemangioblastomas, ovarian cysts, and pheochromocytoma; along with multiple bilateral tumor nodules of clear cell RCC.[8][9]
- Hereditary Leiomyomatosis and Aggressive Papillary Carcinoma Syndrome: is an autosomal dominant disease. There is a mutation of the FH, and associated features include uterine leiomyomatosis. Metastatic spread occurs early in the disease.[9][10]
- Hereditary Papillary Carcinoma: This is also an autosomal dominant form with MET gene mutation. It is manifested by bilateral and multiple papillary tumors. They may have a number of additional cytogenetic abnormalities.[8]
- Birt-Hogg-Dubé syndrome: is an autosomal dominant disease due to mutations involving the BHD gene, which transcribes folliculin. The syndrome manifests a range of histological types for renal tumors. Associated features include fibrofolliculomas, trichodiscomas, and acrochordons. Pulmonary cysts may also be seen.[8][11]
- Tuberous Sclerosis Syndrome: presents with TSC1 and TSC2 gene mutations that transcribe hamartin and tuberin protein. The syndrome manifests with multiple bilateral renal angiomyolipomas and CCRCC along with extra-renal manifestations.[12]
Epidemiology
There had been a steady increase in the incidence of renal cell carcinoma (RCC) since 1975 that has slowed in the past few years. This increase is attributed to the detection of asymptomatic and early cases due to advances and extensive use of imaging techniques. Over half of the RCC cases are detected incidentally.[13] RCC accounts for over 3% of all adult malignancies and has several histological subtypes. The year 2020 estimates suggest that 73,750 cases of renal cancers will be detected (5% of all cancers in males and 3% of all cancers in females), and 14,830 persons will die from the disease. It is a tumor of older age group and is most commonly seen between the ages of 60 to 70 years; it has an approximately 2 to 1 male to female ratio. As opposed to incidence mortality that has reduced by about 1% every year since 2008.[1]
Overall 5-year relative survival in the US is 93% for patients diagnosed in the early stages of the disease. Early disease patients make up about two-thirds of all the cases diagnosed with renal cancer. The overall survival rate for kidney and renal pelvis cancers is 75%.[1]
Pathophysiology
It is generally thought that renal cell carcinomas (RCCs) arise from the epithelial cells of the nephron, linking the CCRCC to the proximal tubular epithelium, PRCC to the distal tubular epithelium and chromophobe RCC (ChRCC) to intercalated cells of collecting duct. However, to the best of our literature search, reliable analysis is lacking. Based on the mice modeling study, it has been proposed that CCRCC may originate in the Bowman capsule.[7][14][15]
Classification of most RCCs on the basis of genetic alterations and molecular signatures is imperfect. However, familial and sporadic VHL gene alterations/mutations are considered as the hallmark of CCRCC.[4][7][9][15][16] In 95% of CCRCC, the loss of short arm of chromosome 3 (3p) has been seen where VHL is located (3p25.3).
The genetic basis of PRCC is mainly based on the inherited form of the disease. The cytogenetic abnormalities associated with the PRCC subtype include trisomy of chromosomes 3, 7, 12, 16, 17, and 20, c-MET mutations and loss of the Y chromosome. PRCC has two main subtypes (see histopathology section). About 15% of nonhereditary PRCC and hereditary PRCC (usually type 1) are characterized by germline MET alteration or gain of chromosome 7. Type 2 PRCC is further grouped based on multiple and distinct genetic alterations in the form of activation of the NRF2-ARE pathway, CDKN2A loss, SETD2 mutations, and TFE3 fusion. A CIMP (CpG island methylator phenotype) suggests a poor prognosis. Hereditary leiomyomatosis associated PRCC have FH mutation and are aggressive tumors.[4][6][7][17]
Chromophobe RCC (ChRCC) has a number of cytogenetic abnormalities associated with it that include loss of multiple chromosomes such as 1, 2, 6, 10, 13, 17, and 21.[18]
Histopathology
Modified 2016 World Health Organization (WHO) classification defines over 14 types of renal cell carcinomas (RCCs):[19]
- Clear cell RCC
- Multilocular cystic renal neoplasm of low malignant potential
- Papillary RCC
- Hereditary leiomyomatosis RCC
- Chromophobe RCC
- Collecting duct carcinoma
- Renal medullary carcinoma
- MiT Family translocation carcinomas
- Succinate dehydrogenase (SDH) deficient RCC
- Mucinous tubular and spindle cell carcinoma
- Tubulocystic RCC
- Acquired cystic disease-associated RCC
- Clear cell papillary RCC
- RCC, unclassified
Major subclasses include clear cell RCC (CCRCC), papillary RCC (PRCC), chromophobe RCC, collecting duct, and unclassified RCC. Xp11 translocation carcinoma is another distinct entity. Only the most common four subtypes will be discussed here.
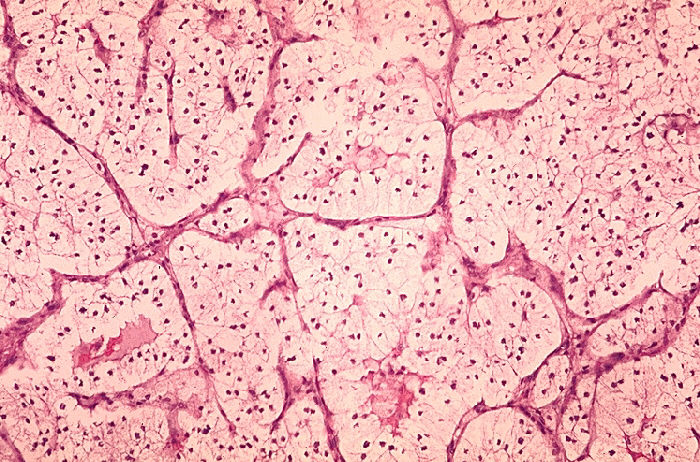
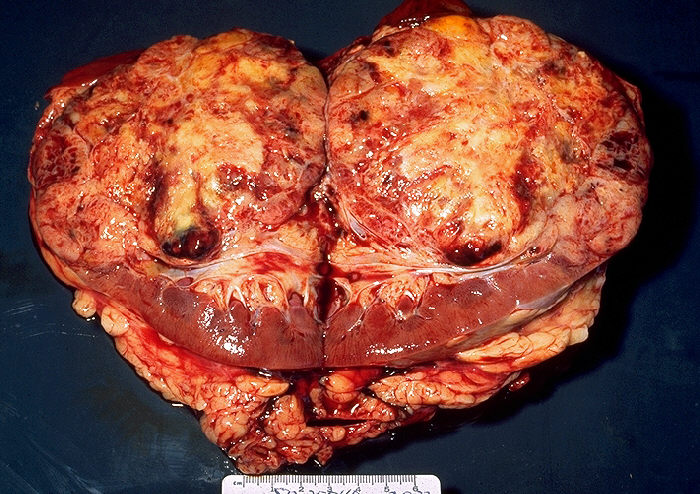
Clear Cell Carcinoma (CCRCC): is the most common type (70-80%) of RCC. Sporadic tumors are usually single and unilateral, unlike the familial lesions, which may be multiple and bilateral. The cut surface is yellow due to large amounts of lipid in the cells, with large areas of necrosis and hemorrhage. The tumor has prominent and delicate vasculature with cystic and solid areas.[17][19] Microscopically the tumor consists of solid to trabecular (cordlike) or tubular (resembling tubules), rarely cystic pattern. The tumor cells are rounded or polygonal and are clear (containing glycogen and lipids) or pink granular cytoplasm (mitochondria rich).[17][19]
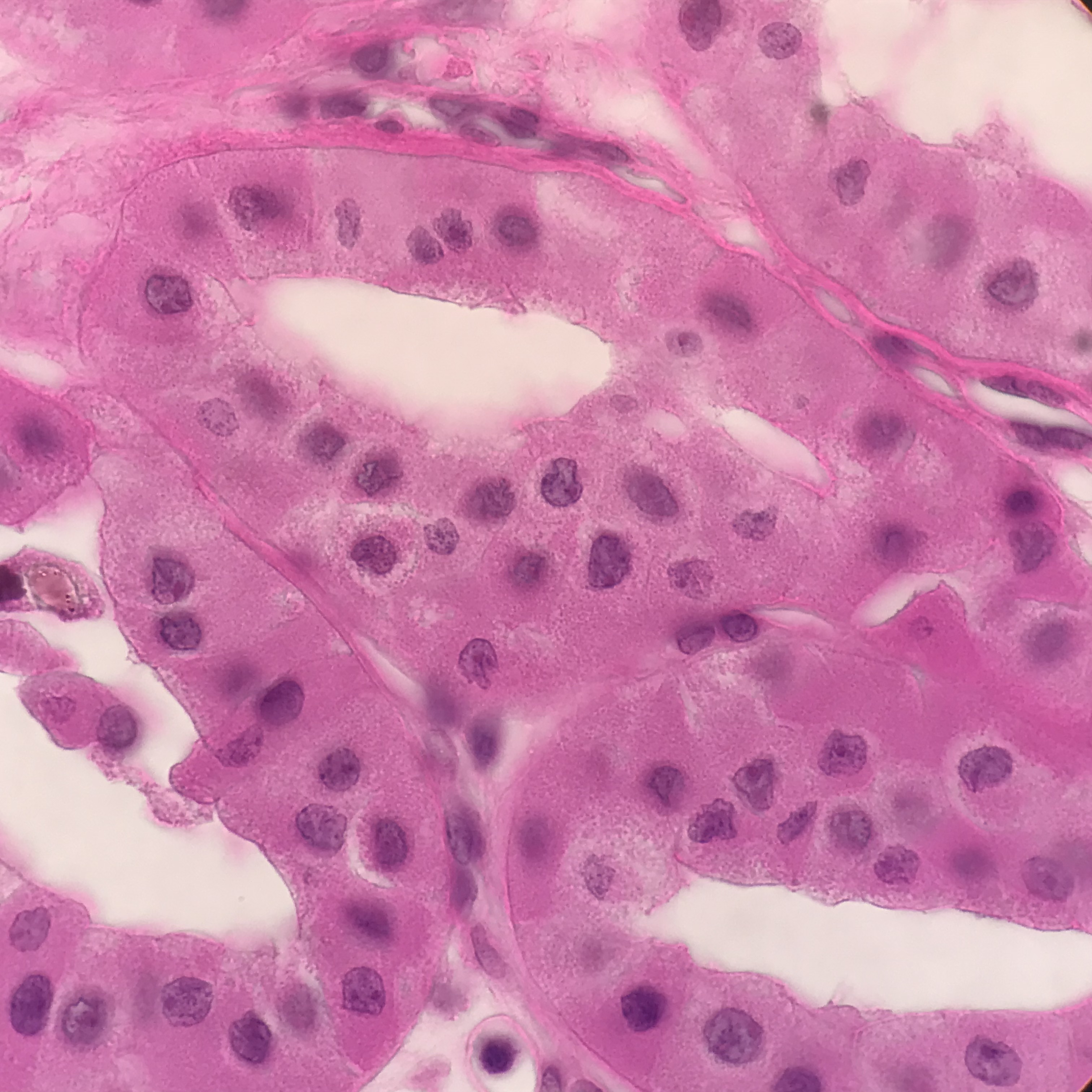
Papillary Carcinoma (PRCC): is about 10-15% of all renal cancers. Small tumors are often incidental findings and can be seen at autopsy. Unlike CCRCC, even sporadic tumors can be multifocal and bilateral. Grossly the tumor is well circumscribed and grayish-white and typically exhibits central necrosis and frequent hemorrhages. Histologically PRCC has two main subtypes. Type 1, are multifocal and characterized by papillae and tubular structures covered with small cells containing basophilic cytoplasm and small, uniform, oval nuclei. Type 2 has a heterogeneous pattern characterized by papillae covered with large cells containing eosinophilic cytoplasm and large, spherical nuclei with prominent nucleoli, suggesting a more aggressive tumor.[17][19]
Chromophobe Carcinoma (ChRCC): constitute about 5% of all RCCs. They are thought to arise from intercalated cells of collecting ducts. The tumor presents as one or more nodules and has a lobulated surface. Tumor cut surface appears orange colored that turns to grayish-white on formalin fixation. The tumor pattern is solid mostly, occasionally cribriform, and may have focal calcifications. Histologically they are made up of basic chromophobe large polygonal cells that have a transparent slightly reticulated cytoplasm and perinuclear halos with thick-walled vasculature. They are difficult to distinguish from oncocytoma.[17][19]
Collecting Duct (Bellini duct) Carcinoma: comprise about 1% or less of RCCs. They arise from cells in the medullary part of collecting ducts. There is no distinct molecular or genetic pattern. Histologically the tumor has irregular channels lined by highly atypical epithelium with a hobnail appearance arranged in glandular pattern enmeshed within a prominent fibrotic stroma. Bellini duct carcinoma needs to be distinguished from medullary carcinoma, which is associated with sickle cell trait.[19][17]
History and Physical
Renal cell carcinomas (RCCs) usually remain asymptomatic until late in the disease, and over 50% of tumors may be detected incidentally.[13] Only 10 to 15% of patients may present with the so-called “classic triad” of flank pain, hematuria and flank fullness. Historically over 60% of patients present with asymptomatic hematuria. Other features include fatigue, weight loss, fever, night sweats, malaise, hypertension, and anemia. Varicocele may develop due to tumor invasion and growth into the renal vein and inferior vena cava that may block the testicular vein. Hypercalcemia, when present, suggests bony metastasis or a paraneoplastic phenomenon.
Bone metastasis is often osteolytic and causes pathologic fractures, spinal cord compression, and hypercalcemia. Excruciating, sharp, band-like back pain may be present due to vertebral collapse and spinal cord compression due to metastatic renal cell carcinoma and may help diagnose metastatic disease.[20] Renal carcinoma is called the “great mimic” because of the frequent occurrence of paraneoplastic syndromes. It can produce a number of substances that lead to hypercalcemia (PTHrP), erythrocytosis (erythropoietin), and Cushing syndrome (ACTH), etc.
A third of the patients with RCC have metastatic disease at presentation. Therefore the physical examination should have an evaluation for metastasis to lungs (75%), bone (20%), liver (18%), CNS (8%), and others.
Evaluation
Initial laboratory investigation in a suspected case of renal cell carcinoma (RCC) should include routine urinalysis with urine cytology,, especially if urothelial tumors are suspected. Assessment of anemia and platelets should be done with a complete blood count and erythrocyte sedimentation rate (ESR). Renal function tests and electrolytes to assess renal insufficiency in order to help make decisions on the use of contrast for radiologic tests.
RCC is frequently associated with paraneoplastic syndromes, which include hypercalcemia, erythrocytosis, and non-metastatic hepatic dysfunction (Stauffer syndrome), etc. Thus, laboratory studies in the evaluation of RCC should include exploration for the paraneoplastic syndromes. Liver function tests help rule out hepatic metastasis and paraneoplastic (non-metastatic) hepatic involvement. Serum calcium and other tests should be included based on the patient’s clinical presentation.
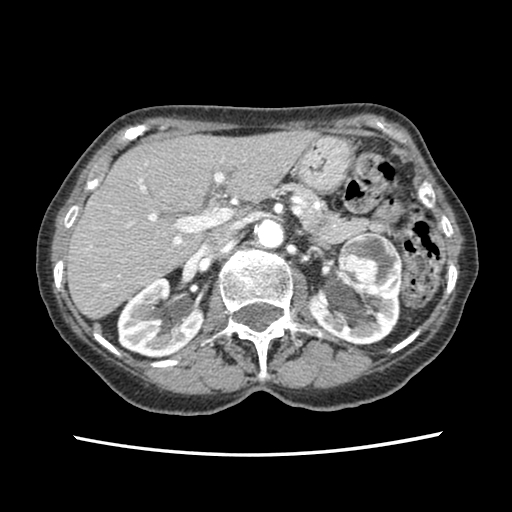
Imaging studies provide both diagnostic confirmation and help plan management. It is important to differentiate between the distinct histologic subtypes of the common RCCs. It is possible to non-invasively discriminate the subtypes based on gross morphologic imaging appearances, signal intensity on T2-weighted magnetic resonance images (MRI), and the degree of tumor enhancement on dynamic contrast-enhanced computed tomography imaging.[21] CCRCC is hypervascular on dynamic contrast-enhanced CT or MRI. Most PRCCs show the low signal intensity and are hypovascular, while ChRCC has a homogeneous solid appearance even when large, and have a central stellate scar and spoke-wheel enhancement.
The tests most commonly utilized are as follows:
- Renal ultrasonography (often the initial study)
- The initial study may include CT excretory urogram.
- Confirmation is done by a staging CT of the abdomen and pelvis.
- Chest X-ray or CT scan for lung metastasis
- Magnetic resonance imaging (MRI) for venous invasion and metastatic tumor progression
- Renal arteriography and venography after RCC have been confirmed, and inferior vena cava involvement is suspected.
- Bone scan and alkaline phosphatase levels to diagnose or assess bone metastasis and follow-up
- CT head, if cerebral metastasis is suspected or clinical picture suggests metastasis.
- MRI may also be used where contrast cannot be used due to patient tolerance
- Urgent magnetic resonance imaging should be performed to rule out cord compression, and high-dose dexamethasone therapy should be started.
- Where the hereditary disease is suspected, either due to the young age at presentation or RCC patients with a family history of the disease, genetic testing for a familial syndrome should be performed.
- Ultrasound or CT guided percutaneous needle biopsy can be done for suspicious lesions and cystic lesions suspected to be malignant.
Biomarker development is a rapidly growing field in oncology as a potential tool for diagnosis and prognosis. Although serum and urinary compounds have been evaluated e.g., TNF, HSP27, serum amyloid A, osteopontin, etc. in serum and nuclear matrix proteins-22, neutrophil gelatinases, aquaporin-1, kidney injury molecule-1, etc. in urine; no serum or urinary biomarker has yet received validation for RCC. Imaging remains the mainstay in RCC for diagnosis, screening, follow-up, and treatment monitoring.
Treatment / Management
The therapeutic options and management of renal cell carcinoma (RCC) are stage-dependent. Therefore accurate staging is essential to effective management (see the section on staging).[21][22]
Stage 1a: Tumors confined to the kidney should be treated with curative intent. Complete surgical resection of the tumor with nephron-sparing partial nephrectomy is recommended. The NCCN recommends that abdominal CT or MRI be performed within six months of surveillance initiation followed by abdominal CT, MRI, or ultrasound at least annually.[22]
Stage 1b: Partial or radical nephrectomy has similar results. Follow-up consists of a baseline abdominal CT, MRI, or ultrasound 3-12 months after nephrectomy, followed annually for three years after partial nephrectomy and at the discretion of the surgeon after radical nephrectomy. Chest X-ray annually for three years.[22]
Stage 2 and 3: Radical nephrectomy with a follow-up schedule has baseline abdominal CT, MRI, or ultrasound abdomen and chest CT after 3 to 6 months and then every 3-6 months for three years, followed by annually up to 5 years. Site-specific imaging as needed.[22]
Stage 4: Systemic targeted molecular therapies have replaced immunotherapy (e.g., interferons-α) using tyrosine kinase inhibitors for a vascular endothelial growth factor (VEGF-TKIs) (e.g., sunitinib, sorafenib, pazopanib, and axitinib) or rapamycin inhibitors for mammalian targets (e.g., temsirolimus and everolimus). Nephrectomy followed by immunotherapy improved survival in patients with metastatic RCC compared to immunotherapy or TKIs alone. Follow-up consists of pretreatment CT or MRI of chest, abdomen, and pelvis, followed by repeat imaging every 6 to 16 weeks, modified by clinical status.[22]
Differential Diagnosis
Nearly 50% of renal cell carcinomas (RCCs) are found incidentally and the disease is generally asymptomatic. Thus, a possibility of RCC should be considered if a renal mass is found on a radiologic study.
The following conditions should be considered in a patient that presents with a renal mass as they can mimic the appearance of RCC:
- Abscess
- Angiomyolipoma (benign neoplasm)
- Renal oncocytoma (benign neoplasm)
- Renal adenoma (benign neoplasm)
- Renal lymphoma
- Renal cyst
- Renal infarction
- Sarcoma
- Metastasis from distant primary lesions e.g. Metastatic melanoma
Differential diagnoses based on the clinical findings would include:
- Acute pyelonephritis
- Bladder cancer
- Chronic pyelonephritis
- Non-Hodgkin lymphoma
- Adult type Wilms tumor
Staging
Tumor Node Metastasis (TNM) classification system, outlined by the American Joint Committee on Cancer (AJCC), is used to stage renal cancer both clinically and pathologically.[23] The AJCC TNM Staging System for renal cancer is detailed in Table 1.
Data taken from the National Cancer Data Base (http://www.facs.org/cancer/ncdb/ index.html) provide the rationale for breaking up T2 into T2a (>7 cm but not more than 10 cm) and T2b (>10 cm). Substantially different outcomes were reported for subgroups. Ipsilateral adrenal involvement has similar outcomes as patients with T4 or M1 disease. Additionally, the presence of multiple adverse features can act collaboratively to further worsen the prognosis. The adverse features include peri-renal fat invasion, tumor size as a continuous variable, size of the largest involved lymph node, and extra-nodal extension. Although, all nodal involvement irrespective of extent suggests a poor prognosis, therefore, nodal involvement is consolidated as N1.
However, tumors with renal vein thrombosis are known to have a relatively favorable prognosis and are therefore staged as T3a rather than T3b. In addition, there are a number of potential molecular prognostic factors, including genetic variables, proliferative markers, angiogenic parameters, growth factors and receptor, and adhesion molecules. Most of these are still not fully validated. Although future staging protocols may capture this information to build individualized management protocols.[23]
TABLE 1: Anatomic stage and Prognostic groups
Stage T N M
I T1 N0 M0
II T2 N0 M0
III T1 or T2 N1 M0
T3 N0 or N1 M0
IV T4 Any N M0
Any T Any N M1
T1: Tumor ≤7 cm in greatest dimension, limited to the kidney, T1a: Tumor ≤4 cm in greatest dimension, limited to the kidney, T1b: Tumor >4 cm but not >7 cm in greatest dimension, limited to the kidney.
T2: Tumor >7 cm in greatest dimension, limited to the kidney, T2a: Tumor >7 cm but ≤10 cm in greatest dimension, limited to the kidney, T2b: Tumor >10 cm, limited to the kidney.
T3: Tumor extends into major veins or perinephric tissues but not into the ipsilateral adrenal gland and not beyond Gerota fascia, T3a: Tumor extends into the renal vein or its segmental branches, or invades the pelvicalyceal system, or invades perirenal and/or renal sinus fat but not beyond Gerota fascia, T3b: Tumor extends into the vena cava below the diaphragm, T3c: Tumor grossly extends into the vena cava above the diaphragm or invades the wall of the vena cava, T4: Tumor invades beyond Gerota fascia (including contiguous extension into the ipsilateral adrenal gland).
Regional Lymph Nodes (N): NX: Regional lymph nodes cannot be assessed, N0: No regional lymph node metastasis, N1: Metastases in regional lymph node(s).
Distant Metastasis (M): M0: No distant metastasis, M1: Distant metastasis.
Prognosis
The 5-year relative survival rate for RCC is 75%. Two-thirds of cases are diagnosed at a local stage, for which the 5-year relative survival rate is 93%.[1] Prognosis is best predicted by stage and grade with cancer-specific survival rates that approximate 85-90% for clinically localized (stage 1 and 2) RCC. The International Society of Urological Pathology (ISUP) Grading System was proposed in 2012 and later adopted by WHO. WHO/ISUP system for renal cell carcinoma includes more objective criteria for tumor grading based on nuclear characteristics. Specific grading factors include the degree of tumor invasion, the presence and extent of venous thromboembolic involvement, adrenal gland involvement, tumor grade, sarcomatoid features-if present, the lymphovascular invasion, and the extent of necrosis.[24] Chromophobe RCC is no longer graded in the ISUP system. In general, the higher grade is associated with larger tumor size and more aggressive tumors.
These prognostic factors can be grouped into three main categories that are tumor-related factors, patient-related factors, and laboratory biochemical tests. The use of these factors to create integrative algorithms improves the prognostic power over a simple anatomic tumor stage alone and improve management decisions.[23]
Prognostic features for RCC
- Tumor related: Stage, tumor size, tumor grade, histologic type, histologic tumor necrosis, sarcomatoid transformation
- Patient-related: Asymptomatic vs. local symptoms vs. systemic symptoms, performance status, substantial weight loss, presence of well-defined paraneoplastic syndrome, metastasis-free interval, history of prior nephrectomy
- Laboratory biochemical tests: Elevated LDH levels, hypercalcemia, anemia, thrombocytosis, elevated ESR, or CRP.
Complications
Possible complications in RCC may include:
- Direct tumor effects e.g., hypertension, constipation, abdominal bloating, etc.
- Paraneoplastic phenomenon e.g., erythrocytosis, hypercalcemia, non-metastatic liver involvement e.g., Stauffer syndrome characterized by high levels of serum alkaline phosphatase, erythrocyte sedimentation rate, α-2-globulin, and γ-glutamyl transferase. Thrombocytosis, prolonged prothrombin time, and hepatosplenomegaly may be seen.
- Metastasis e.g., pulmonary function compromise (pulmonary metastasis), varicocele (venous spread), bony pain and spinal cord compression (bone metastasis), seizures (brain metastasis), jaundice and transaminase elevation (liver metastasis).
- Adverse effects from targeted systemic therapies or surgery may include renal function compromise, hypertension, proteinuria, impaired wound healing, gastrointestinal perforation, hemorrhage and thrombosis, reversible leukoencephalopathy, impaired cardiac function and endocrine dysfunction e.g., hypothyroidism.
Pearls and Other Issues
Renal cell carcinoma (RCC) accounts for over 3% of all malignancies and 90% of adult renal malignancies. It is the most lethal of all urologic cancers. The 5-year survival rate varies based on histologic type as does metastatic potential and favored site of metastasis with the lung being the most frequently involved in clear cell tumors and the liver in chromophobe tumors.
Tobacco use and obesity are the strongest risk factors, and about half of the RCC can be eliminated by controlling these two factors.
RCC is a group of closely related neoplasms; the subtypes can be separated on the basis of histologic findings, cytogenetic abnormalities, biologic behavior, and imaging appearances of the tumors. It is possible to differentiate the three main subtypes (clear cell, papillary, and chromophobe) based on characteristic radiologic appearances.
Histopathologically, RCC has a number of subtypes, and clear cell (70%), papillary (10%), and chromophobe (5%) are the most common. Other RCC subtypes are rare and include carcinoma of the collecting ducts of Bellini (1%), renal medullary carcinoma, Xp11.2 translocation carcinoma, among others.
Cytogenetically familial and sporadic VHL gene alterations/mutations are seen in CCRCC with a loss of 3p25.3 locus. PRCC has chromosomal abnormalities of 3, 7, 12, 16, 17, and 20, c-MET mutations, and loss of the Y chromosome. Chromophobe RCC has a number of abnormalities that include loss of multiple chromosomes such as 1, 2, 6, 10, 13, 17, and 21.
Management and prognosis are dependent mainly on the staging of the tumor, supplemented by tumor grading and special characteristics like the degree of tumor invasion, the presence and extent of venous thromboembolic involvement, adrenal gland involvement, tumor grade, sarcomatoid features-if present, the lymphovascular invasion, and the extent of necrosis.
Tumors (< 7 cm) confined to one pole of the kidney are treated by partial nephrectomy, and kidney confined tumors can be treated by partial or radical nephrectomy. Targeted tyrosine kinase inhibitors (TKIs) e.g., sunitinib, which is antiangiogenic, improves median progression-free survival. Novel immunotherapeutic agents (T-cell checkpoint inhibitors) show promise in improving outcomes in metastatic RCC.
Enhancing Healthcare Team Outcomes
Renal cell carcinoma (RCC) is a relatively common tumor; however, with early detection and effective management, the 5-year survival rates are excellent (over 90%). The majority of patients first present to the primary care physician or internist, and if suspected, urologist consultation should be sought early. The key to improved survival is early diagnosis and prompt referral. Stringent adherence to follow-up protocols will catch recurrence early for effective and proper management. The oncology nurse and social worker should be involved in effective post-operative care and follow up as well as assist the provider in monitoring the patient and provide regular follow-up support. The pharmacist needs to educate the patient for drug effects, side effects, and complications of targeted immunotherapy. Thus, an interprofessional team approach is necessary for patients’ physical and mental health and support and positive outcomes as the recovery and follow-ups are rigorous and prolonged over five or more years.