Continuing Education Activity
Rotor syndrome, also known as Rotor type hyperbilirubinemia, is an autosomal recessive disease and a rare cause of mixed direct (conjugated) and indirect (unconjugated) hyperbilirubinemia. The disease is characterized by non-hemolytic jaundice due to chronic elevation of predominantly conjugated bilirubin. This phenomenon results from impaired hepatocellular storage of conjugated bilirubin that leaks into plasma, causing hyperbilirubinemia. Its presenting symptom is jaundice, but Rotor syndrome is a benign, self-limiting disorder that does not require treatment. This activity describes the pathophysiology and presentation of rotor syndrome and highlights the role of the interprofessional team in its management.
Objectives:
- Describe the pathophysiology of Rotor syndrome.
- Outline the presentation of a patient with Rotor syndrome.
- Summarize the treatment for Rotor syndrome.
- Explain the importance of improving care coordination among interprofessional team members to improve outcomes for patients affected by Rotor syndrome.
Introduction
Rotors syndrome (also known as Rotor type hyperbilirubinemia) is an autosomal recessive disease and a rare cause of mixed direct (conjugated) and indirect (unconjugated) hyperbilirubinemia. The disease is characterized by non-hemolytic jaundice due to chronic elevation of predominantly conjugated bilirubin (over 50%). This phenomenon results from impaired hepatocellular storage of conjugated bilirubin that leaks into plasma, causing hyperbilirubinemia. Its presenting symptom is jaundice, but Rotor Syndrome is a benign and self-limiting disorder that does not require treatment.[1][2] Generally, patients are asymptomatic, and jaundice is usually an incidental finding. Regarding symptoms, it is similar to Dubin-Johnson syndrome; however, the liver is histologically normal.
As opposed to Dubin-Johnson syndrome, liver hyperpigmentation is absent in Rotor syndrome. In addition, the serum bilirubin in Gilbert syndrome is mostly unconjugated, unlike in Rotor syndrome. Rotor syndrome generally begins shortly after birth or during childhood. Jaundice could be intermittent, and conjunctival icterus could be the only clinical manifestation. The manifestation of jaundice/icterus is because of conjugated hyperbilirubinemia, excess coproporphyrin in urine, and near-absent liver uptake of anionic diagnostics, including cholescintigraphic tracers.[3]
Etiology
The exact etiology of Rotor syndrome (RS) is unknown but appears to be attributable to a deficiency in the intra-cellular capacity of the liver to store organic anions, such as bilirubin diglucuronide.[4] Rotor syndrome is an autosomal recessive disorder caused by homozygous mutations in both the SLCO1B1 and SLCO1B3 genes on chromosome 12. These genes provide instructions for making organic anion-transporting polypeptides 1B1 and 1B3 (OATP1B1 and OATP1B3, respectively). These proteins are found in liver cells and mediate sodium-independent cellular uptake of compounds, including bilirubin glucuronide, bile acids, steroid and thyroid hormones, numerous drugs, toxins, and their conjugates. They share several structural features, such as similar 12 transmembrane domains with intracellularly situated N- and C-termini.[5]
In a normal liver, a majority of bilirubin is conjugated by hepatocytes and secreted back into the blood. It is then reabsorbed in downstream hepatocytes by the OATP1B1 and OATP1B3 proteins.[6] In Rotor syndrome, the OATP1B1 and OATP1B3 proteins are abnormally short; therefore, the bilirubin is less efficiently taken up by the liver and removed from the body, causing a buildup of bilirubin in the blood and urine, which results in jaundice and dark urine.[7][8][9][10]
A study of eight families of Rotor syndrome found that it was associated with mutations that are predicted to cause deficiencies of the organic anion transporters OATP1B1 and OATP1B3. These crucial detoxification-limiting proteins carry out the uptake and clearance of a large number of drugs and drug conjugates through the sinusoidal hepatocyte membrane. The observation suggested the risk of life-threatening toxicities with the use of certain medications in Rotor syndrome.[1]
A recent study looked at the insertion of long interspersed nuclear element-1 (LINE-1) in genes potentially causing Rotor syndrome.[11]
Epidemiology
Rotor syndrome was first described in 1948 by Rotor et al. in the Philippines. Since then, cases have been reported worldwide, including in the US, Japan, France, Mexico, Papua New Guinea, and Italy. The exact prevalence of the condition is not known. As most patients are asymptomatic, the condition may be picked up based on incidental findings on investigations done for other reasons. It is the second rare hereditary cause of hyperbilirubinemia, the first being Crigler-Najjar type 1.[12] Furthermore, there is no gender predisposition seen with Rotor syndrome. While the disease often presents in adolescence or early adulthood, it has been reported to present shortly after birth or during childhood.[2][13]
Pathophysiology
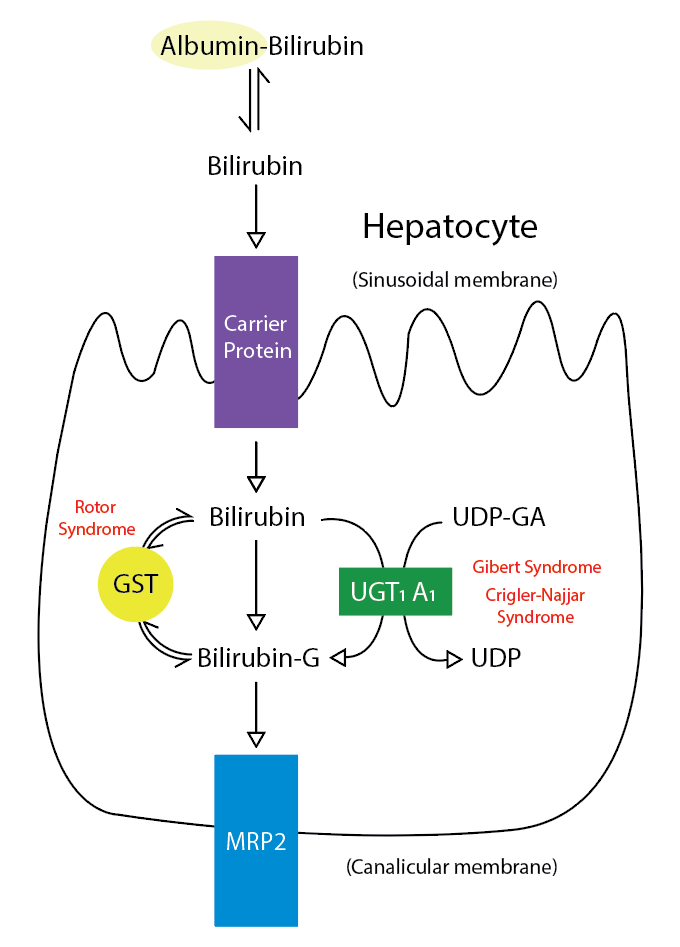
As mentioned, Rotor syndrome is caused by mutations in two proteins responsible for transporting bilirubin and other compounds from the blood to the liver to be metabolized and cleared from the body. Coproporphyrin 1, a significant coproporphyrin isomer in bile, is transported from the hepatocyte back into circulation and is excreted in the urine. Thus, urine coproporphyrin is elevated in Rotor syndrome. Cholescintigraphy using sulfobromophthalein (BSP) has shown that the transport capacity of dye into bile is reduced by less than 50%, and the storage capacity in the hepatocytes is decreased more than five-fold compared with normal values in this disease.[2][9]
The normal disposition of bilirubin includes hepatic transport through an albumin carrier, where it is conjugated with glucuronic acid to bilirubin glucuronic acid, a water-soluble compound that is easy to excrete in the feces. The donor compound is uridine diphosphate glucuronic acid, and the catalyst of the reaction is uridine diphosphate-glucuronyltransferase (UGT1A1-specific isoform). A reduction in the activity of this enzyme leads to the development of Gilbert syndrome and Crigler-Najjar syndrome (types 1 and 2). Glutathione-S-transferase acts as an intracellular carrier for certain organic molecules. Patients with rotor syndrome may also have a deficiency in hepatic glutathione-S-transferase.[14] A deficiency results in abnormal uptake of bilirubin within the cytosol. Additionally, bilirubin conjugates are bound to glutathione-S-transferase in the process of secretion from the liver cells into the canalicular lumen. Deficiencies in intracellular storage lead to the leakage of these conjugates back into circulation.[15]
Rotor syndrome is inherited as an autosomal recessive disorder. In one study, it was found to be coexistent with other inherited disorders, such as glucose-6-phosphate dehydrogenase (G6PD) deficiency and heterozygous beta-thalassemia, which suggests a potential interaction between co-inherited genes.[16]
The defective storage and excretion of organic anions, such as bilirubin, lead to impaired secretion of conjugated bilirubin, which leads to its reabsorption into the bloodstream and subsequent excretion in the urine. The resulting hyperbilirubinemia manifests as jaundice. As opposed to Dubin-Johnson syndrome, hepatic pigmentation has not been reported.
Histopathology
A liver biopsy is not required to diagnose Rotor syndrome, but if done, a liver biopsy in patients with the disease reveals normal histology. Liver biopsy may help distinguish Rotor syndrome from other, more serious liver diseases. Since Rotor syndrome is clinically similar to Dubin-Johnson syndrome (DJS), it is imperative to distinguish between these two conditions; the absence of dark melanin-like pigments on liver biopsy distinguishes Rotor Syndrome from Dubin-Johnson syndrome.[13][17][18]
History and Physical
Patients with Rotor syndrome are typically asymptomatic; however, generalized non-pruritic jaundice can present at birth or early childhood. These symptoms may come and go. Some patients only have scleral icterus. Patients also complain about passing dark-colored urine, long-term jaundice, and fatigue. Additionally, around 5% to 30% of patients may also experience abdominal pain, gastric mucosal abnormalities, and fever.[2][17]
Physical examination is typically normal, except for mild jaundice. Excess bilirubin results in a yellow-orange tinge of the tissues that most easily appears as icteric (yellowish) discoloration of the sclera. Unlike other disorders with cholestasis, pruritus is absent. Another feature to remember in Rotor syndrome is that there will be no hepatosplenomegaly in the presence of jaundice which can help narrow down the list of differentials.
Based on some distinguishing features, Rotor syndrome can be differentiated from other similar disorders, such as Dubin–Johnson syndrome. For instance, the liver has black pigmentation in Dubin–Johnson syndrome, while in Rotor syndrome liver has normal histology and appearance.
Evaluation
Rotor syndrome is largely a diagnosis of exclusion. Serological abnormalities in Rotor syndrome only include elevated total serum bilirubin (typically elevated between 2 to 5 mg/dL but may be as high as 20 mg/dL). Most of the time, alanine aminotransferase, aspartate aminotransferase, gamma-glutamyl transferase, and alkaline phosphatase levels are normal, but mild elevations can be seen. If any of these lab values are markedly elevated, investigation for other, more serious conditions is warranted. Imaging studies cannot diagnose Rotor syndrome but can help rule out other diseases that cause hyperbilirubinemia. For example, an ultrasound of the liver and biliary tree can help investigate the causes of extra-hepatic biliary obstruction. The gallbladder is visualized on oral cholecystography in Rotor syndrome, while it is not visualized in DJS.[19] Ultimately, the best method of diagnosing the disease is the analysis of urine coproporphyrin excretion. The total urine coproporphyrin excretion in Rotor syndrome has a two to five-fold elevation, with 65% constituting coproporphyrin 1.[2][17]
- Total coproporphyrin excretion is greatly elevated in both Rotor syndrome and DJS:
- The ratio of coproporphyrin 1 to 3 in urine allows the differentiation of these two conditions.
- Around 90% is coproporphyrin 1 in Dubin-Johnson syndrome but a much lower proportion in Rotor syndrome.
- The plasma disappearance of injected bromosulfophthalein is delayed, with no secondary rise (which is seen in Dubin-Johnson syndrome).
- The hepatic biopsy will show pigment deposition in Dubin-Johnson syndrome but not Rotor syndrome.[2]
Treatment / Management
Rotor syndrome is a benign disease requiring no treatment. Jaundice is a lifelong finding, but the disease is not associated with morbidity or mortality, and life expectancy is not affected. Most individuals with Rotor syndrome are born to consanguineous couples, and its diagnosis may coincidently identify consanguinity. Distinguishing Rotor syndrome from other more serious disorders is important to avoid unnecessary workup and interventions. It is also critical to reassure and calm patients or family members of patients with Rotor syndrome that the condition is benign.[2][9][13][20]
Differential Diagnosis
The many causes of hyperbilirubinemia can be divided into causes of unconjugated versus conjugated hyperbilirubinemia and then further classified. Some conditions that can cause hyperbilirubinemia to include:
- Dubin-Johnson syndrome
- Gilbert syndrome
- Crigler-Najjar syndrome (type 1 and type 2)
- Extra-hepatic biliary obstruction
- Familial intra-hepatic cholestasis
- Benign recurrent intrahepatic cholestasis (BRIC)
- Drug-induced hepatotoxicity
- Hemolysis
- Cholestasis of pregnancy
- Viral hepatitis
- Autoimmune hepatitis
- Wilson disease
- Hemochromatosis
- Alpha-1-antitrypsin deficiency
- Cirrhosis
It is important to differentiate Rotor syndrome from other diseases causing hyperbilirubinemia. Normal alkaline phosphatase levels and gamma-glutamyltranspeptidase help distinguish Rotor syndrome from disorders associated with biliary obstruction. Abnormal urinary coproporphyrin excretion and normal liver histology help distinguish this entity from DJS.[18][17]
Prognosis
Rotor syndrome is a benign condition and persists for life. There are no recommended management options for patients suffering from Rotor syndrome. There is no morbidity or mortality associated with Rotor syndrome; hence its prognosis is good. Mortality and morbidity occur if there is another coexisting liver disease.
Complications
Rotor syndrome is a benign disease with no effect on life expectancy. No adverse drug effects have been documented in people with Rotor syndrome, but the absence of the hepatic proteins OATP1B1 and OATP1B3 may lead to serious issues with liver uptake. OATP1B1 plays a role in drug detoxification, and with reduced activity of this protein, certain drugs such as anticancer agents, methotrexate, and statins can accumulate and result in drug toxicity.[21] Caution should be taken before administering these drugs.[2]
Deterrence and Patient Education
There are no specific recommendations for patients suffering from Rotor syndrome; however, patients should be given reassurance that the disease is benign and that the jaundice is not associated with progressive liver damage.
Pearls and Other Issues
Making a correct and early diagnosis of Rotor syndrome is crucial, as misdiagnoses of this disease often lead to a more costly, lengthy, and invasive workup that can place a patient at risk of avoidable complications and financial burden. As this disease is generally diagnosed in the pediatric population, it is important to reassure concerned family members that Rotor syndrome is benign with no effect on life expectancy and the occasional appearance of jaundice.
Enhancing Healthcare Team Outcomes
Rotor syndrome is a benign disorder with no morbidity and is best managed by an interprofessional team that includes clinicians (MDs, DOs, NPs, and PAs), pharmacists, and nurses. The efforts of these interprofessional team members revolve around the differential diagnosis, as described above, as the syndrome requires no therapeutic interventions. Meticulous record-keeping and open communication among team members will lead to an accurate diagnosis and the best outcomes.
A liver biopsy is not required to diagnose Rotor syndrome, but if done, a liver biopsy in patients with the disease reveals normal histology. In addition, a liver biopsy may help distinguish Rotor syndrome from other, more severe liver diseases. Since Rotor syndrome is clinically similar to Dubin-Johnson syndrome (DJS), it is imperative to differentiate between these two conditions; the absence of dark melanin-like pigments on liver biopsy distinguishes Rotor syndrome from DJS.
The outcomes for patients with Rotor syndrome are excellent.