Introduction
Inheritance patterns illustrate disease transmission amongst generations of a family. An autosomal pattern of inheritance occurs in families affected with a genetic disease whose gene is not on a sex chromosome. Patients affected with autosomal recessive (AR) diseases have a disease allele on each chromosome. The pattern of individuals affected with an AR disease can be traced through a family to determine which individuals are carriers and which individuals are likely to become impacted.
Development
Mendelian genetics focuses on disorders caused by single genes, thus excluding polygenic or multifactorial traits. Mendelian genetics has its basis on the experiments of Gregor Mendel, the Austrian monk who worked on garden peas. Mendel described three laws.[1] The first law is the Law of Segregation, which states that each gamete contains one allele for a trait, and it combines with the other gamete to give the offspring a pair of alleles for each trait, one from each parent. The second law is the Law of Independent Assortment, which states that alleles for different traits may segregate and combine independently of each other during meiosis and fertilization. The third law is the Law of Dominance, which states that one allele can be dominant to the other, thereby masking the other if both are present. The masked allele, in this case, is known as the recessive allele.
Mechanism
Diseases transmitted in an autosomal recessive pattern present when an individual inherits two recessive disease alleles; thus, the individual will be affected with the disease phenotype. Following Mendel’s Law of Segregation, this means that both parents must have at least one disease allele that they can pass on to their child. The easiest way to determine the inheritance pattern of a disorder in a family is by looking at a pedigree. Autosomal recessive diseases typically affect both females and males equally. Autosomal recessive patterns manifest by skipping generations as the affected are usually children of unaffected carriers. It is also common to see affected individuals with unaffected offspring. This type of pattern demonstrates horizontal transmission because the affected individuals are often seen in more than one child in a family and not in all generations (the affected individuals trace a horizontal line through the pedigree).
In contrast, when a family has affected individuals in every generation, this is considered vertical transmission (the affected individuals trace multiple vertical lines through the pedigree). Vertical transmission is characteristic of an autosomal dominant disease. Additionally, consanguinity in a family affected with an autosomal recessive condition will often lead to children afflicted with the condition.[2]
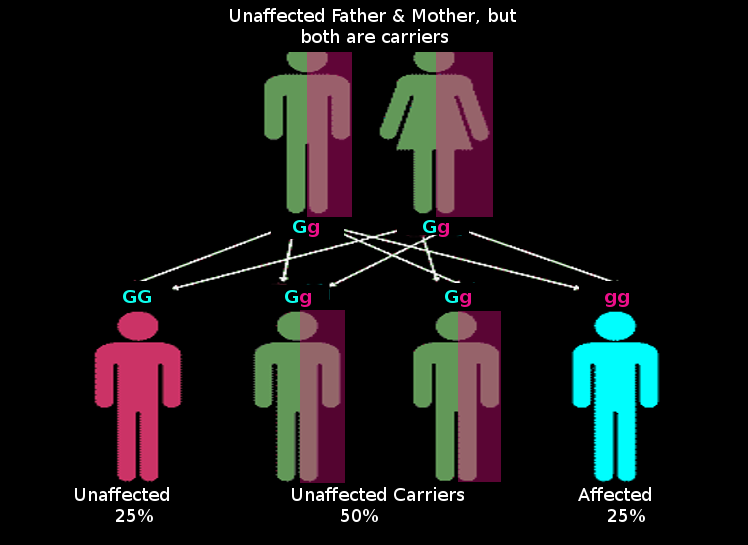
The most common situation of an autosomal recessive disease occurs when the parents are each carrier or heterozygous (Dd). Children of carrier parents have a 25% chance of inheriting the disorder. This value is obtained by using the Punnet square model used in genetics. Each parent has a 50% chance of passing on the disease allele. Using the multiplication rule of probability, there is a 50% chance that the father passes on his disease allele and a 50% chance that the mother passes on her disease allele; 50% x 50% = 25%. So with the mating of carrier parents, there is a 25% chance that the child will be affected, a 50% chance that the child would be a carrier, and 25% chance that they would be homozygous dominant and unaffected.
An exception to the horizontal transmission seen in most cases of autosomal recessive disorders occurs in the case of pseudodominance. In these cases, there will be affected individuals in more than one generation.[3] Pseudodominance occurs when a carrier parent (Dd) has a child with an affected person (dd). In this case, there is a 50% chance that the child will be affected, and there is a vertical transmission pattern on a pedigree; this is distinguishable from autosomal dominant traits by tracing the pedigree and looking for autosomal recessive characteristics elsewhere in the pedigree. These characteristics include affected parents who have unaffected children and unaffected parents who have affected children.
Testing
Physical exams and laboratory tests can help to diagnose many autosomal recessive disorders so that counseling, management, and treatment can commence immediately. Obtaining a thorough family medical history is an integral part of the diagnostic process, as the clinical phenotype and inheritance pattern can provide essential information about which genes require testing and which technology to use.
The field of genetics is rapidly growing as we understand that more and more diseases have an underlying genetic cause. With services that guide parents on their reproductive risks, the medical field is improving the non-clinical realm of treatment options. However, understanding and education are still lacking. In the case of autosomal recessive disorders, consanguinity proves to be one of the most significant risks.[4] Health care providers should encourage genetic counseling for their patients, especially patients from highly consanguineous communities such as those in the Middle East, Ashkenazi Jews, etc.
Clinical Significance
Many disorders are inherited in an autosomal recessive manner. Some of the more well-known disorders include cystic fibrosis, Tay-Sachs disease, sickle cell disease, and thalassemia.[5]
Cystic fibrosis (CF) is an autosomal recessive disorder that is caused by a mutation in the CFTR gene located on chromosome 7. This systemic illness occurs in about 1 in 1000 White births, and about 1 in every 30 people carry a disease allele.[6] CF affects the exocrine glands, including the pancreas, sweat, and salivary glands, and causes an inability to secrete chloride at the surface of cells.[7] This often results in patients with lung infections, pancreatic insufficiencies, and even infertility. Carriers of this disorder are phenotypically unaffected because the amount of protein produced from one functional allele provides sufficient chloride ion transport through the apical membranes at the cell surface. In contrast, patients have two disease alleles.
Tay-Sachs disease is another well-known autosomal recessive early-onset disorder that results from a mutation in the HEXA gene. The HEXA gene encodes the protein hexosaminidase A, which is present in lysosomes. A deficiency of hexosaminidase A causes a buildup of gangliosides in the brain and thus results in brain and spinal damage. Similar to cystic fibrosis, carriers of this disorder are phenotypically unaffected. One wild type HEXA allele can produce enough hexosaminidase enzyme to cleave the gangliosides in the brain. These individuals are therefore considered to be haplo-sufficient, where half the amount of enzyme activity is enough. Individuals who have two disease alleles are affected. Babies with this disorder often cannot hold their heads up, may have dementia, a startled response, blindness, and even early death.[8] This is most prevalent in the Ashkenazi Jewish population, French Canadians, and Louisiana Cajuns.
Hemoglobinopathies are also relatively frequent AR disorders. Sickle cell disease and alpha and beta-thalassemia affect hemoglobin and its oxygen-carrying capacity. In patients with sickle cell disease, the mutation causes hemoglobin to be a different shape that is not as conducive to carrying oxygen. These hemoglobin molecules polymerize into long chains that give the red blood cells their sickle shape.[9] These red blood cells do not have the same deformability as normal red blood cells and thus get stuck in small blood vessels. Affected individuals display characteristic traits such as splenomegaly, anemia, and swelling of limbs due to occluded blood vessels. In patients with thalassemia, the relative production of the alpha and beta-globin chains is altered, which impairs the assembly of the hemoglobin molecule; thus, there are not enough molecules to carry oxygen.[10] These individuals characteristically present with microcytic anemia, shortness of breath, hepatosplenomegaly.[11]
Sickle cell disease also exhibits incomplete dominance and haploinsufficiency. Individuals who carry a single disease allele (heterozygotes) have an intermediate trait (sickle cell trait) that is phenotypically somewhere between unaffected (homozygous normal allele) and severely affected (homozygous disease allele giving rise to sickle cell anemia). These individuals are not considered completely unaffected, hence incomplete dominance. Hemoglobinopathies are most prevalent among African populations and subtropical populations where malaria is rampant because the carriers of these genes have protection against malaria.[12]