Specimen Requirements and Procedure
Principles of Western Blotting
The principles of western blotting are equal loading of proteins, separation of proteins by molecular weight, electrophoretic transfer to a suitable membrane, and probing of antibodies.
Equal Loading of Proteins
Proper sample preparation for subsequent electrophoresis is crucial for downstream analysis. Western blot samples are first prepared by protein extraction with specialized cell lysis buffers and protease and phosphatase inhibitors (PPIs). There are numerous extraction methods, and proper selection is determined by the sample type. For example, most tissue preparation is by homogenization or sonication; however, osmotic shock or detergent lysis is more suited for easily lysed cells such as erythrocytes or cultured cells. Furthermore, the cell lysis buffer used in extraction should align with the target protein cellular localization.[8] For example, radioimmunoprecipitation assay buffer (RIPA) is more adept for nuclear and mitochondrial proteins.
Although rare, some antibodies will not be able to detect denatured samples. As such, gentle buffers without detergents are required. PPIs are used to maintain the structure and phosphorylation status of the target protein from the activity of endogenous phosphatases upon cell lysis and exogenous phosphatases in the lysis microenvironment. Collectively, this information underscores the need to tailor protein extraction to the sample type and the target protein.[2]
There must be an equal concentration of proteins per western blot sample. Intuitively, this is imperative for a valid experiment, as unequal proteins per lane can skew the analysis. Protein concentration is quantifiable by conducting a Bradford assay, a colorimetric protein assay that exploits the interaction of a dye with a protein.[9] In brief, the dye Coomassie Brilliant Blue G-250 complexes with proteins to change color, and this absorbance shift gets recorded by a spectrophotometer. Thus, by running this assay with known protein standards, a linear regression standard curve is generated to calculate unknown protein extract concentrations in the sample.[10]
All western blot samples have three elements: protein extract, cell lysis buffer, and Laemmli (sample) buffer. Protein extract is normalized with cell-lysis buffer to the desired protein concentration, and an equal volume of Laemmli (sample) buffer is added. Therefore, a western blot sample always has a 1-to-1 volume ratio of normalized protein and Laemmli buffer. Laemmli buffer (60 mM Tris-HCl pH 6.8; 20% glycerol; 2% SDS; 4% beta-mercaptoethanol; 0.01% bromophenol blue) is unique to western blot sample preparation as each reagent is purposeful for SDS-PAGE.[11] Glycerol adds density to samples, so they drift into the loading wells.
Bromophenol blue (BPB) is a nonreactive reagent that serves as a dye front for electrophoresis. Sodium dodecyl sulfate (SDS) is a potent anionic detergent that coats denatured proteins with an equal anion-to-mass ratio; this masks proteins' charge, shape, and size characteristics and renders them solely a function of molecular weight.[12] Beta-mercaptoethanol (BME) is a reducing agent that acts on disulfide bonds; without BME, proteins with disulfide bonds retain some shape and do not electrophorese consummately by molecular weight.[13] Tris-HCl pH 6.8 works with the discontinuous buffer system, explained in further detail below. Prepared samples are heated before loading to further denature proteins to their primary structure. Thus, proteins undergo electrophoresis by their monomeric weight.[14]
Separation of Proteins by Molecular Weight
The separation of proteins by weight is possible due to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) due to its combinatory use of a detergent and a discontinuous buffer system.
Typically, PAGE is an analytical biochemistry method used to separate contents such as nucleic acids and proteins by electrophoretic mobility in a chemically inert gel; however, by adding SDS, a potent anionic detergent, all denatured proteins will be coated with an equal charge to mass ratio. Therefore, the rate of protein migration is proportional to weight. Indeed, larger proteins travel slower than smaller proteins due to retarding properties of the porous gel. A gel matrix is formed from the polymerization of acrylamide and the crosslinking of N, N'-methylenebisacrylamide. This matrix creates a molecular sieve that imbues retarding properties. The pore size of this sieve is alterable by adjusting the percentages of polyacrylamide and N, N'-methylenebisacrylamide as they are inversely proportional.[15]
Two different-sized sieves are used in PAGE: a stacking gel and a resolving gel. As the name suggests, the stacking gel stacks proteins into a narrow band to allow proteins to enter the resolving gel simultaneously, which is made possible due to its bigger pore size and acidity. With its much smaller pore size and basicity, the resolving gel is where the separation of proteins occurs.[16]
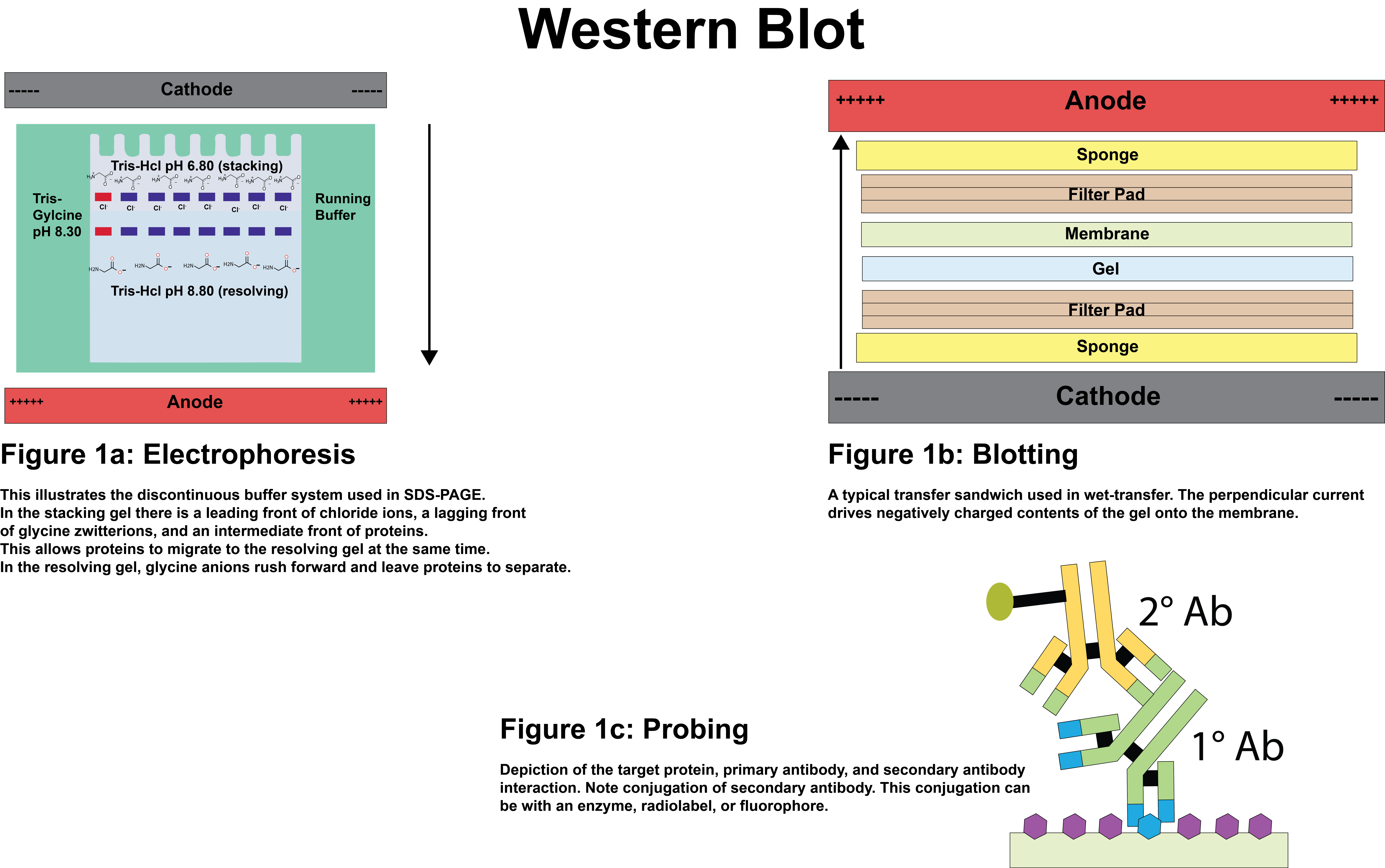
The Laemmli discontinuous buffer system is most commonly used in SDS-PAGE. This system utilizes running buffer (25 mM Tris; 192 mM glycine; 0.1% SDS; pH~8.30) as electrode buffer and tris-HCl to buffer an acidic stacking gel (pH~6.80) and a basic resolving gel (pH~8.80). The deliberate use of varied pHs exploits the charge properties of glycine.[17] In an acidic environment, glycine is a zwitterion, but in a basic environment, it is a glycinate anion. Thus, when electrophoresis starts, the current quickly draws glycinate into the stacking gel. The acidic gel protonates glycine to its zwitterionic form, thereby severely impeding its mobility.[18]
In contrast, chloride from the tris-HCl buffer disassociates from its counter ion and migrates quickly to the anode. Proteins lie between a trailing front of glycine and a leading front of chloride; this results in all proteins arriving at the resolving gel simultaneously, a vital component for subsequent separation.[19] The basicity of the resolving gel reforms conjugate glycinate anions at the stacking-resolving gel interface. From this interface, glycinate anions quickly migrate past the protein front. The proteins now hit the resolving gel in narrow bands without a zone of high voltage previously formed from the leading and trailing ions in the stacking gel.[16] Thus, this allows proteins to migrate down the resolving gel slower, which induces the separation of proteins due to the higher concentration of polyacrylamide (Figure 1a).
The samples run in their respective lanes alongside a molecular weight marker, often called a protein ladder. For example, a typical setup would have the ladder in the first lane and the samples in the remaining lanes. The ladder establishes standard molecular weight bands that are then used to read the relative weight of proteins.[20]
Electrophoretic Transfer (Blotting)
Blotting is the electrophoretic transfer of gel contents onto a suitable membrane; in a western blot, the contents are proteins. There are multiple methods of blotting in addition to numerous types of membranes. Although various transfer systems exist (wet, semi-dry, fast), the main principle of electrophoretic transfer remains the same. Like electrophoresis, negatively charged samples migrate toward an anode; in blotting, a transfer sandwich with a slightly modified electrode buffer is used. Towbin buffer (25 mM Tris; 192 mM glycine; 20% methanol; pH 8.3) is the standard transfer buffer, although small tweaks to this buffer are possible for the target protein.[21]
Methanol is important in blotting as it increases the hydrophobicity of proteins and facilitates the release of SDS, both of which increase the adsorption of proteins onto the membrane. From cathode to anode, the sandwich organizes as filter paper, polyacrylamide gel, membrane, and filter paper.[22] In a wet transfer system, fiber pads or sponges are placed superficially on each side. The sandwich is subjected to a perpendicular current that drives gel contents onto the membrane. (Figure 1b).
Equilibration of sandwich contents in transfer buffer is crucial for increasing transfer efficiency; it prevents the drying of both the gel and membrane, washes electrophoretic contaminants off the gel, and reforms the original gel size.[23] As electrophoresis runs, voltage increases temperatures, and this increases gel size. Thus cold transfer buffer shrinks gels to the proper size. Interestingly, methanol in the transfer buffer also cools the gel during equilibration.[24]
Each transfer system has advantages, and selecting one largely depends on the target protein and lab workflow. Among various transfer apparatuses, the two most commonly used are wet and semi-dry.[22] It is essential to consider the main differences between wet and semi-dry systems: the volume of transfer buffer used and transfer time. Wet transfer uses a tank transfer system that requires a large volume of transfer buffer, whereas semi-dry transfer systems typically require only the dampening of the sandwich.[25]
Semi-dry systems are also time-efficient as blotting usually finishes within an hour, but a low voltage gets applied overnight in a wet transfer. While semi-dry transfer seems to be the better option as there is a significant reduction in both the volume of transfer buffer and length of transfer time, it has its limitations.[26] Large proteins like membrane receptors do not blot well, and overall transfer efficiency is lower. Wet transfer shines in its ability to yield high efficiency across a wide range of protein sizes, thus offering the most flexibility.[27]
When Drs. Burnette and Towbin published their seminal studies, electrophoretic transfer was carried out on nitrocellulose membranes. They remained the gold standard until the advent of polyvinylidene difluoride (PVDF) membranes. Concisely, PVDF membranes outcompete nitrocellulose membranes in their protein binding capacity, chemical resistance, and enhanced transfer efficiency in the presence of SDS.[28] PVDF promotes higher adsorption of proteins, and its chemical resistance allows for the stripping and reprobing of membranes. Also, transfer efficiency markedly improves by inserting a small percentage of SDS in the transfer buffer. However, noted protein sensitivity from PVDF could also increase the background signal for analysis.[29]
Methanol in transfer buffer can shrink nitrocellulose membranes and precipitate out large proteins. Both membranes come in different pore sizes, and membrane pore size is directly related to protein weight.[6] Smaller proteins require smaller pore sizes, although a pore size of 0.45 microns is suitable for most proteins.[30] Recent years have seen the development of unique membranes such as those used for near-infrared detection systems. As such, the type of membrane chosen should reflect the target protein and downstream detection systems.[6]
Antibody Probing
Upon completing the electrophoretic transfer, proteins are now on the membrane, and two antibodies serve for probing and analysis. The primary antibody that binds a specific region on the target protein is used to detect its presence on the membrane. The secondary antibody conjugates with a component used for analysis.[31] This antibody indirectly binds the target protein by binding to the constant regions of the primary antibody (Figure 1c).
Since membranes have a high affinity for protein, before probing, membranes are incubated in a buffer to coat the remaining surface area. This ‘blocking’ buffer includes a protein with a minimal binding affinity to the target protein and, consequently, the antibody. Typically, blocking buffer proteins include either casein from powdered milk or bovine serum albumin (BSA). Although casein is cheaper and suitable for most proteins, BSA is a better choice when the target protein is phosphorylated; there is a cross-reactivity between casein and phosphorylation-specific primary antibodies.[30] After blocking, the membrane is washed with TBS-T, a mixture of tris-buffered saline, and tween 20. Tween 20 is a nonionic detergent that helps remove peripherally bound proteins on the membrane.[32]
Probing of primary and secondary antibodies is done by incubating the membrane in a probing buffer of either the primary or secondary antibody in TBS-T. The membrane is first incubated in the primary probing buffer, typically overnight in a cold room, and washed again with TBS-T.[33] The membrane is then incubated with the secondary probing buffer for about an hour, then washed. These washing steps are crucial to reduce background noise in the analysis. After probing and washing, the membrane is ready to be read.[32]
As mentioned earlier, the secondary antibody conjugates with a component specific to the type of analysis. Autoradiography was a common way to visualize bands but has declined in its popularity due to hazards associated with this method. It uses a radiolabeled isotope conjugated to the secondary antibody.[34] More commonly, a chemiluminescence method is used. This method uses substrates that react with an enzyme-conjugated secondary antibody. These enzymes are horseradish peroxidase (HRP) or alkaline phosphatase (AP).[35]
The enzyme-mediated reaction produces light that is then recorded with an imaging system. More recently, secondary antibodies have been conjugated with fluorophores that can be detected without the need for substrates.[7] This fluorescence-based detection is gaining popularity due to its capability of probing two target proteins via secondary antibodies with different wavelength fluorophores; this is a selective advantage for relative protein expression analysis as housekeeping proteins are visible alongside a protein of interest.[6]
The visualization of bands can serve different analytical purposes. Simply, the presence of bands can verify the expression of a protein, whereas the density of bands can show comparative relative protein expression. A housekeeping protein is also probed to evaluate relative protein expression.[36] A housekeeping protein is a ubiquitous protein constitutively expressed in all cells. By normalizing the band densities of the target protein with those of the housekeeping protein, a statistically significant difference between sample types can be measured.[37]
Clinical Significance
As highlighted earlier, a western blot has a considerable amount of steps. This lengthy process drives up the time and cost needed for accurate results. However, unlike an ELISA, the western blot is less likely to give false-positive results, especially in diagnosing human immunodeficiency virus (HIV).[41]
Western blotting is used to detect anti-HIV antibodies in human serum and urine samples.[42] The protein samples from a known HIV-infected individual get separated by electrophoresis and then blotted on the nitrocellulose membrane. Then a specific antibody is affixed to detect the protein.[43] The western blot is usually performed after the ELISA test to confirm the diagnosis of HIV. It is far more sensitive than the ELISA test.[44] More recently, in commercial HIV western blot kits, viral proteins come affixed to the membrane. Antibodies from human urine or serum samples bind to these proteins, and anti-HIV antibodies are used to detect bands alongside quality controls.[45]
The western blot is also useful in detecting Lyme disease and atypical and typical bovine spongiform encephalopathy.[46][47]
The western blotting procedure determines the presence or absence, size, and abundance of target proteins in a sample which is beneficial for various scientific reasons across many fields of study.[48] In this regard, the technique proves beneficial to evaluate the following: protein-DNA interactions, protein-protein interactions, post-translational modifications (PTMs), protein isoform detection, antibody characterization, epitope mapping, and subcellular protein localization.[38]
As western blotting is an antibody-based method, the technique often supports and produces reliable results for detecting non-infectious diseases using high throughput screening. For example, when determining cancers, incongruous isoforms of proteins can become potential markers of the pathology of the disease.[49] Autoantibodies may also indicate an autoimmune disease.[38]
Some proteins are engineered, through molecular cloning, to contain short sequences of amino acids that serve as a tag. Common tags include the HA-tag and the Myc-tag. These tags serve as a foreign protein epitope that does not naturally occur in the biological system being studied.[50] Thus, the tag makes the protein easier to detect than all other naturally occurring proteins. An antibody directed to the tag will identify the presence and amount of the tagged protein in the western blot.[7]
Quality Control and Lab Safety
Quality Controls
Like any experiment, quality controls should be used to validate findings. In a western blot, a positive control, negative control, loading control, and a no first-degree A-B control are all effective in achieving and maintaining robust experiments.[49] Controls are dedicated lanes wherein the sample is altered specifically for the control type. A positive control is a sample known to contain the target protein, whereas a negative control is known not to contain the target protein. This can be as general as different organ types or as specific as different cellular localization. For example, if analysis of the expression of a nuclear protein is the aim, and subcellular fractioning is done to isolate this region, a negative control evaluates the quality of fractioning, nonspecific binding of antibodies, and a false-positive.[38]
Positive controls are powerful in verifying that the workflow is well-optimized, even in the absence of bands in sample lanes. Also, a positive control can verify a negative result.[51] Loading control is a housekeeping protein such as alpha-tubulin or beta-actin.[49] Probing with antibodies specific for a housekeeping protein checks for an equal amount of proteins per sample. A nonspecific secondary antibody can yield false positives. The specificity of a secondary antibody is evaluated by not incubating a membrane strip with the primary antibody.[38]
Troubleshooting
Unfortunately, this method has many error arms due to many steps and a lengthy workflow. Discussing every possible error, its cause, and the solution is outside the scope of this review. Instead, the most common issues and how to troubleshoot them are described below.
Smiling of Bands
When bands are not migrating equally down the gel, this pattern can exaggerate to a smiley pattern. This indicates that the gel has air bubbles, the voltage is too high, or the volume of the loading sample is too large.[7] Air bubbles within the gel can distort the migration of bands. A constant voltage during electrophoresis is directly proportional to resistance, and since resistance and temperature are directly linked, a high voltage increases the temperature in the electrophoresis tank.[52] Heat pockets and an overall increase in the temperature of the running buffer can also alter migration. Before the buffer can warm up, a high voltage at the start of electrophoresis will rush bands and cause nonlinear migration. A large volume of loading samples can cause spillover into other lanes, and these large bands can skew into another lane.[49]
Absence of Bands
If the detection system shows no signal across all lanes except the ladder, there are many possible causes. It is best first to localize where in the workflow the error occurred. Typically, the most common culprits are poor transfer efficiency or poor probing.[7]
By staining the membrane with Ponceau S, a membrane-safe red dye, bands can be visualized. If bands are well illustrated on the membrane, particularly where the target protein is expected, it indicates that transfer efficiency is not likely the cause. If there are no bands, transfer settings must be altered.[51] A washout of proteins can occur in which the proteins from the membrane migrate to the filter paper. This is because the transfer time is too high; reducing either voltage or transfer time can prevent washout.[7]
A poor transfer can also occur if little to no proteins are adsorbed on the membrane. To understand the directionality of transfer, the gel can be stained to reveal bands.[49] A significant visualization of bands can suggest the actual transfer was poor rather than a high voltage or time. Rechecking the quality of the transfer buffer, increasing transfer settings, and ensuring proper contact between the gel and membrane can resolve this issue. If the target protein is small, a semi-dry system may be preferable.[38]
If a positive control lane is used and bands are absent, this can be due to a poor detection kit, poor antibodies, or even an incorrect antibody concentration. Antibody concentration is optimized by running titration experiments.[7]
Multiple Bands
Only a single row of bands should be visualized in detection. The presence of multiple bands suggests the nonspecific binding of antibodies.[52] Polyclonal antibodies typically produce this result, as well as a high antibody concentration. As mentioned earlier, titration experiments should be performed to optimize detection.[7]
High Background With or Without Splotches
Poor membrane blocking, excessive antibody concentration, and a dry membrane can result in high background signals. Increasing the period for blocking or changing the type of protein used in blocking buffer may solve this.[49] Titration experiments have to be run for antibody optimization. Insufficient washing can result in high background signal, a significant cause of splotches on the membrane. Membranes must be maintained wet throughout the experiment, and a dry membrane can give high background signals.[38]
Lab Safety
Standard lab safety rules apply. When casting gels, acrylamide is a potent neurotoxin; however, it is chemically inert once polymerized. The careful handling of this reagent is necessary, and proper precautionary measures should be met before handling it.[53]