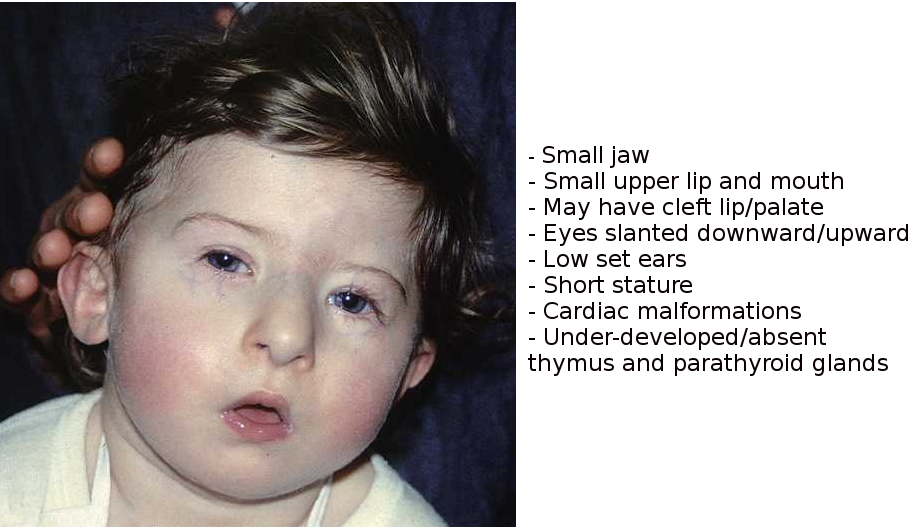
[1]
McDonald-McGinn DM, Sullivan KE, Marino B, Philip N, Swillen A, Vorstman JA, Zackai EH, Emanuel BS, Vermeesch JR, Morrow BE, Scambler PJ, Bassett AS. 22q11.2 deletion syndrome. Nature reviews. Disease primers. 2015 Nov 19:1():15071. doi: 10.1038/nrdp.2015.71. Epub 2015 Nov 19
[PubMed PMID: 27189754]
[2]
Fomin AB, Pastorino AC, Kim CA, Pereira CA, Carneiro-Sampaio M, Abe-Jacob CM. DiGeorge Syndrome: a not so rare disease. Clinics (Sao Paulo, Brazil). 2010:65(9):865-9
[PubMed PMID: 21049214]
[3]
Cascella M, Muzio MR. Early onset intellectual disability in chromosome 22q11.2 deletion syndrome. Revista chilena de pediatria. 2015 Jul-Aug:86(4):283-6. doi: 10.1016/j.rchipe.2015.06.019. Epub 2015 Sep 8
[PubMed PMID: 26358864]
[4]
Cioffi S, Martucciello S, Fulcoli FG, Bilio M, Ferrentino R, Nusco E, Illingworth E. Tbx1 regulates brain vascularization. Human molecular genetics. 2014 Jan 1:23(1):78-89. doi: 10.1093/hmg/ddt400. Epub 2013 Aug 14
[PubMed PMID: 23945394]
[5]
Paylor R, Glaser B, Mupo A, Ataliotis P, Spencer C, Sobotka A, Sparks C, Choi CH, Oghalai J, Curran S, Murphy KC, Monks S, Williams N, O'Donovan MC, Owen MJ, Scambler PJ, Lindsay E. Tbx1 haploinsufficiency is linked to behavioral disorders in mice and humans: implications for 22q11 deletion syndrome. Proceedings of the National Academy of Sciences of the United States of America. 2006 May 16:103(20):7729-34
[PubMed PMID: 16684884]
[6]
Grati FR, Molina Gomes D, Ferreira JC, Dupont C, Alesi V, Gouas L, Horelli-Kuitunen N, Choy KW, García-Herrero S, de la Vega AG, Piotrowski K, Genesio R, Queipo G, Malvestiti B, Hervé B, Benzacken B, Novelli A, Vago P, Piippo K, Leung TY, Maggi F, Quibel T, Tabet AC, Simoni G, Vialard F. Prevalence of recurrent pathogenic microdeletions and microduplications in over 9500 pregnancies. Prenatal diagnosis. 2015 Aug:35(8):801-9. doi: 10.1002/pd.4613. Epub 2015 Jun 24
[PubMed PMID: 25962607]
[7]
Botto LD, May K, Fernhoff PM, Correa A, Coleman K, Rasmussen SA, Merritt RK, O'Leary LA, Wong LY, Elixson EM, Mahle WT, Campbell RM. A population-based study of the 22q11.2 deletion: phenotype, incidence, and contribution to major birth defects in the population. Pediatrics. 2003 Jul:112(1 Pt 1):101-7
[PubMed PMID: 12837874]
[8]
McDonald-McGinn DM, Minugh-Purvis N, Kirschner RE, Jawad A, Tonnesen MK, Catanzaro JR, Goldmuntz E, Driscoll D, Larossa D, Emanuel BS, Zackai EH. The 22q11.2 deletion in African-American patients: an underdiagnosed population? American journal of medical genetics. Part A. 2005 Apr 30:134(3):242-6
[PubMed PMID: 15754359]
[9]
Zhang Z, Huynh T, Baldini A. Mesodermal expression of Tbx1 is necessary and sufficient for pharyngeal arch and cardiac outflow tract development. Development (Cambridge, England). 2006 Sep:133(18):3587-95
[PubMed PMID: 16914493]
[10]
Guner-Ataman B, González-Rosa JM, Shah HN, Butty VL, Jeffrey S, Abrial M, Boyer LA, Burns CG, Burns CE. Failed Progenitor Specification Underlies the Cardiopharyngeal Phenotypes in a Zebrafish Model of 22q11.2 Deletion Syndrome. Cell reports. 2018 Jul 31:24(5):1342-1354.e5. doi: 10.1016/j.celrep.2018.06.117. Epub
[PubMed PMID: 30067987]
[11]
Sumitomo A, Horike K, Hirai K, Butcher N, Boot E, Sakurai T, Nucifora FC Jr, Bassett AS, Sawa A, Tomoda T. A mouse model of 22q11.2 deletions: Molecular and behavioral signatures of Parkinson's disease and schizophrenia. Science advances. 2018 Aug:4(8):eaar6637. doi: 10.1126/sciadv.aar6637. Epub 2018 Aug 15
[PubMed PMID: 30116778]
Level 3 (low-level) evidence
[12]
Meechan DW, Maynard TM, Tucker ES, Fernandez A, Karpinski BA, Rothblat LA, LaMantia AS. Modeling a model: Mouse genetics, 22q11.2 Deletion Syndrome, and disorders of cortical circuit development. Progress in neurobiology. 2015 Jul:130():1-28. doi: 10.1016/j.pneurobio.2015.03.004. Epub 2015 Apr 9
[PubMed PMID: 25866365]
[13]
McDonald-McGinn DM, Sullivan KE. Chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). Medicine. 2011 Jan:90(1):1-18. doi: 10.1097/MD.0b013e3182060469. Epub
[PubMed PMID: 21200182]
[14]
Kruszka P, Addissie YA, McGinn DE, Porras AR, Biggs E, Share M, Crowley TB, Chung BH, Mok GT, Mak CC, Muthukumarasamy P, Thong MK, Sirisena ND, Dissanayake VH, Paththinige CS, Prabodha LB, Mishra R, Shotelersuk V, Ekure EN, Sokunbi OJ, Kalu N, Ferreira CR, Duncan JM, Patil SJ, Jones KL, Kaplan JD, Abdul-Rahman OA, Uwineza A, Mutesa L, Moresco A, Obregon MG, Richieri-Costa A, Gil-da-Silva-Lopes VL, Adeyemo AA, Summar M, Zackai EH, McDonald-McGinn DM, Linguraru MG, Muenke M. 22q11.2 deletion syndrome in diverse populations. American journal of medical genetics. Part A. 2017 Apr:173(4):879-888. doi: 10.1002/ajmg.a.38199. Epub
[PubMed PMID: 28328118]
[15]
Al-Sukaiti N, Reid B, Lavi S, Al-Zaharani D, Atkinson A, Roifman CM, Grunebaum E. Safety and efficacy of measles, mumps, and rubella vaccine in patients with DiGeorge syndrome. The Journal of allergy and clinical immunology. 2010 Oct:126(4):868-9. doi: 10.1016/j.jaci.2010.07.018. Epub
[PubMed PMID: 20810153]
[16]
Bakare N, Menschik D, Tiernan R, Hua W, Martin D. Severe combined immunodeficiency (SCID) and rotavirus vaccination: reports to the Vaccine Adverse Events Reporting System (VAERS). Vaccine. 2010 Sep 14:28(40):6609-12. doi: 10.1016/j.vaccine.2010.07.039. Epub 2010 Jul 30
[PubMed PMID: 20674876]
[17]
McGhee SA, Lloret MG, Stiehm ER. Immunologic reconstitution in 22q deletion (DiGeorge) syndrome. Immunologic research. 2009:45(1):37-45. doi: 10.1007/s12026-009-8108-7. Epub
[PubMed PMID: 19238335]
[18]
Janda A, Sedlacek P, Hönig M, Friedrich W, Champagne M, Matsumoto T, Fischer A, Neven B, Contet A, Bensoussan D, Bordigoni P, Loeb D, Savage W, Jabado N, Bonilla FA, Slatter MA, Davies EG, Gennery AR. Multicenter survey on the outcome of transplantation of hematopoietic cells in patients with the complete form of DiGeorge anomaly. Blood. 2010 Sep 30:116(13):2229-36. doi: 10.1182/blood-2010-03-275966. Epub 2010 Jun 7
[PubMed PMID: 20530285]
Level 3 (low-level) evidence
[19]
Davies EG, Cheung M, Gilmour K, Maimaris J, Curry J, Furmanski A, Sebire N, Halliday N, Mengrelis K, Adams S, Bernatoniene J, Bremner R, Browning M, Devlin B, Erichsen HC, Gaspar HB, Hutchison L, Ip W, Ifversen M, Leahy TR, McCarthy E, Moshous D, Neuling K, Pac M, Papadopol A, Parsley KL, Poliani L, Ricciardelli I, Sansom DM, Voor T, Worth A, Crompton T, Markert ML, Thrasher AJ. Thymus transplantation for complete DiGeorge syndrome: European experience. The Journal of allergy and clinical immunology. 2017 Dec:140(6):1660-1670.e16. doi: 10.1016/j.jaci.2017.03.020. Epub 2017 Apr 8
[PubMed PMID: 28400115]
[20]
Markert ML, Devlin BH, Chinn IK, McCarthy EA. Thymus transplantation in complete DiGeorge anomaly. Immunologic research. 2009:44(1-3):61-70. doi: 10.1007/s12026-008-8082-5. Epub
[PubMed PMID: 19066739]