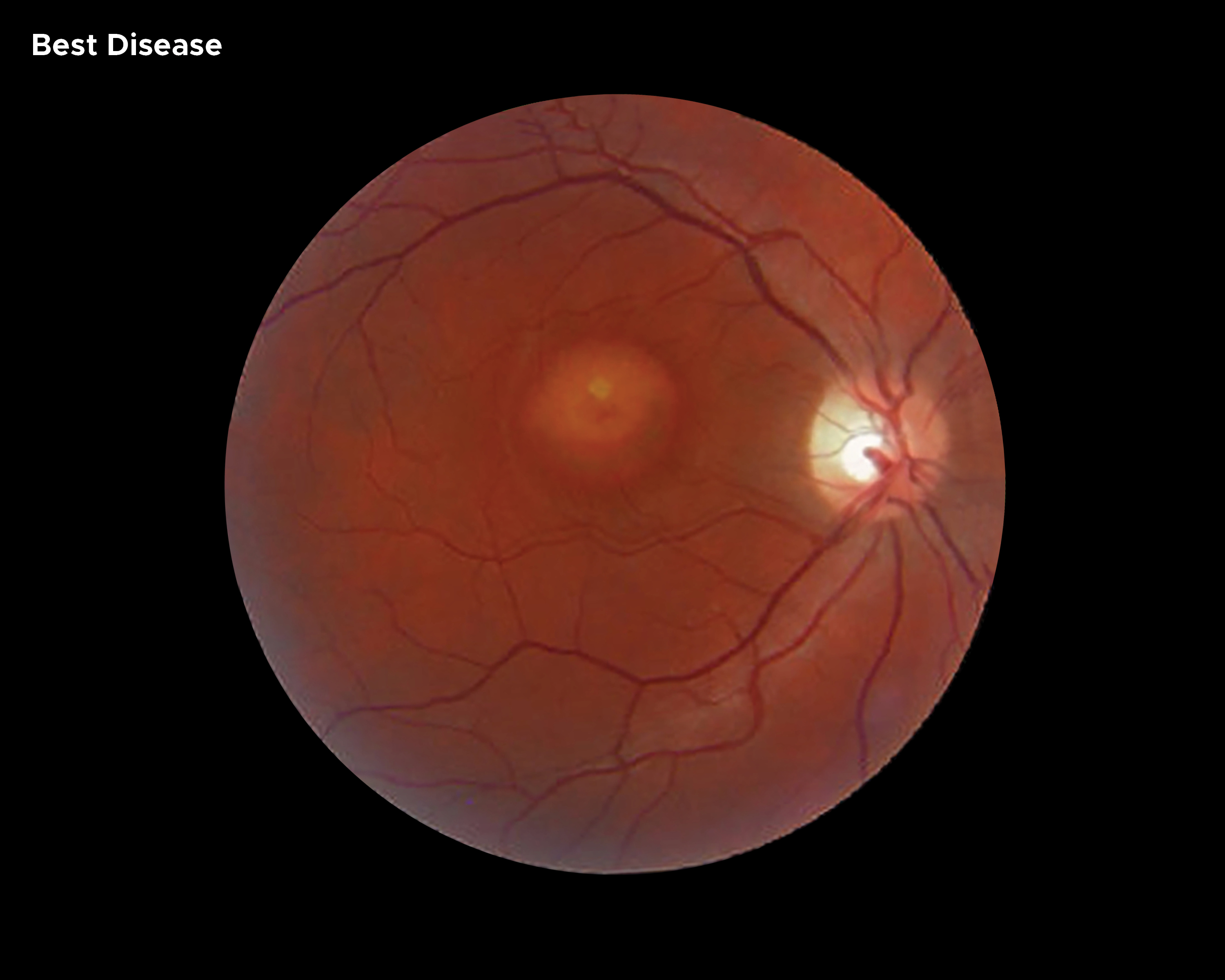
[1]
Zhao L,Grob S,Corey R,Krupa M,Luo J,Du H,Lee C,Hughes G,Lee J,Quach J,Zhu J,Shaw PX,Kozak I,Zhang K, A novel compound heterozygous mutation in the BEST1 gene causes autosomal recessive Best vitelliform macular dystrophy. Eye (London, England). 2012 Jun;
[PubMed PMID: 22422030]
[2]
Bitner H,Mizrahi-Meissonnier L,Griefner G,Erdinest I,Sharon D,Banin E, A homozygous frameshift mutation in BEST1 causes the classical form of Best disease in an autosomal recessive mode. Investigative ophthalmology
[PubMed PMID: 21467170]
[3]
Iannaccone A,Kerr NC,Kinnick TR,Calzada JI,Stone EM, Autosomal recessive best vitelliform macular dystrophy: report of a family and management of early-onset neovascular complications. Archives of ophthalmology (Chicago, Ill. : 1960). 2011 Feb;
[PubMed PMID: 21320969]
[4]
Querques G,Zerbib J,Santacroce R,Margaglione M,Delphin N,Rozet JM,Kaplan J,Martinelli D,Delle Noci N,Soubrane G,Souied EH, Functional and clinical data of Best vitelliform macular dystrophy patients with mutations in the BEST1 gene. Molecular vision. 2009 Dec 31;
[PubMed PMID: 20057903]
[5]
Stone EM,Nichols BE,Streb LM,Kimura AE,Sheffield VC, Genetic linkage of vitelliform macular degeneration (Best's disease) to chromosome 11q13. Nature genetics. 1992 Jul;
[PubMed PMID: 1302019]
[6]
Lin Y,Li T,Ma C,Gao H,Chen C,Zhu Y,Liu B,Lian Y,Huang Y,Li H,Wu Q,Liang X,Jin C,Huang X,Ye J,Lu L, Genetic variations in Bestrophin‑1 and associated clinical findings in two Chinese patients with juvenile‑onset and adult‑onset best vitelliform macular dystrophy. Molecular medicine reports. 2018 Jan;
[PubMed PMID: 29115605]
[7]
Johnson AA,Guziewicz KE,Lee CJ,Kalathur RC,Pulido JS,Marmorstein LY,Marmorstein AD, Bestrophin 1 and retinal disease. Progress in retinal and eye research. 2017 May;
[PubMed PMID: 28153808]
[8]
Mullins RF,Oh KT,Heffron E,Hageman GS,Stone EM, Late development of vitelliform lesions and flecks in a patient with best disease: clinicopathologic correlation. Archives of ophthalmology (Chicago, Ill. : 1960). 2005 Nov;
[PubMed PMID: 16286623]
[9]
Dalvin LA,Pulido JS,Marmorstein AD, Vitelliform dystrophies: Prevalence in Olmsted County, Minnesota, United States. Ophthalmic genetics. 2017 Mar-Apr;
[PubMed PMID: 27120116]
[11]
Tripathy K. Choroidal neovascular membrane in intraocular tuberculosis. GMS ophthalmology cases. 2017:7():Doc24. doi: 10.3205/oc000075. Epub 2017 Sep 1
[PubMed PMID: 28944155]
Level 3 (low-level) evidence
[12]
O'Gorman S,Flaherty WA,Fishman GA,Berson EL, Histopathologic findings in Best's vitelliform macular dystrophy. Archives of ophthalmology (Chicago, Ill. : 1960). 1988 Sep;
[PubMed PMID: 3415551]
[13]
Zhang Q,Small KW,Grossniklaus HE, Clinicopathologic findings in Best vitelliform macular dystrophy. Graefe's archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie. 2011 May;
[PubMed PMID: 21136072]
[14]
Weingeist TA,Kobrin JL,Watzke RC, Histopathology of Best's macular dystrophy. Archives of ophthalmology (Chicago, Ill. : 1960). 1982 Jul;
[PubMed PMID: 7092654]
[15]
Frangieh GT,Green WR,Fine SL, A histopathologic study of Best's macular dystrophy. Archives of ophthalmology (Chicago, Ill. : 1960). 1982 Jul;
[PubMed PMID: 7092655]
[16]
Bakall B,Radu RA,Stanton JB,Burke JM,McKay BS,Wadelius C,Mullins RF,Stone EM,Travis GH,Marmorstein AD, Enhanced accumulation of A2E in individuals homozygous or heterozygous for mutations in BEST1 (VMD2). Experimental eye research. 2007 Jul;
[PubMed PMID: 17477921]
[17]
Arora R,Khan K,Kasilian ML,Strauss RW,Holder GE,Robson AG,Thompson DA,Moore AT,Michaelides M, Unilateral BEST1-Associated Retinopathy. American journal of ophthalmology. 2016 Sep;
[PubMed PMID: 27287821]
[18]
Pece A,Gaspari G,Avanza P,Magni R,Brancato R, Best's multiple vitelliform degeneration. International ophthalmology. 1992 Nov;
[PubMed PMID: 1490837]
[19]
Duncker T,Greenberg JP,Ramachandran R,Hood DC,Smith RT,Hirose T,Woods RL,Tsang SH,Delori FC,Sparrow JR, Quantitative fundus autofluorescence and optical coherence tomography in best vitelliform macular dystrophy. Investigative ophthalmology
[PubMed PMID: 24526438]
[20]
Jarc-Vidmar M,Kraut A,Hawlina M, Fundus autofluorescence imaging in Best's vitelliform dystrophy. Klinische Monatsblatter fur Augenheilkunde. 2003 Dec;
[PubMed PMID: 14704944]
[21]
Tripathy K,Chawla R,Mittal K,Temkar S, Egg yolk in the eye: an ultrawide field evaluation. BMJ case reports. 2016 Jan 27
[PubMed PMID: 26818692]
Level 3 (low-level) evidence
[22]
Ferrara DC,Costa RA,Tsang S,Calucci D,Jorge R,Freund KB, Multimodal fundus imaging in Best vitelliform macular dystrophy. Graefe's archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie. 2010 Oct;
[PubMed PMID: 20414784]
[23]
Battaglia Parodi M,Iacono P,Romano F,Bandello F, SPECTRAL DOMAIN OPTICAL COHERENCE TOMOGRAPHY FEATURES IN DIFFERENT STAGES OF BEST VITELLIFORM MACULAR DYSTROPHY. Retina (Philadelphia, Pa.). 2018 May;
[PubMed PMID: 28376040]
[24]
Khan KN,Mahroo OA,Islam F,Webster AR,Moore AT,Michaelides M, FUNCTIONAL AND ANATOMICAL OUTCOMES OF CHOROIDAL NEOVASCULARIZATION COMPLICATING BEST1-RELATED RETINOPATHY. Retina (Philadelphia, Pa.). 2017 Jul;
[PubMed PMID: 27764019]
[25]
Sodi A,Murro V,Caporossi O,Passerini I,Bacci GM,Caputo R,Menchini U, Long-Term Results of Photodynamic Therapy for Choroidal Neovascularization in Pediatric Patients with Best Vitelliform Macular Dystrophy. Ophthalmic genetics. 2015 Jun;
[PubMed PMID: 25675349]
[26]
Yannuzzi NA,Mrejen S,Capuano V,Bhavsar KV,Querques G,Freund KB, A Central Hyporeflective Subretinal Lucency Correlates With a Region of Focal Leakage on Fluorescein Angiography in Eyes With Central Serous Chorioretinopathy. Ophthalmic surgery, lasers
[PubMed PMID: 26431298]
[27]
Mohler CW,Fine SL, Long-term evaluation of patients with Best's vitelliform dystrophy. Ophthalmology. 1981 Jul;
[PubMed PMID: 7267039]