Introduction
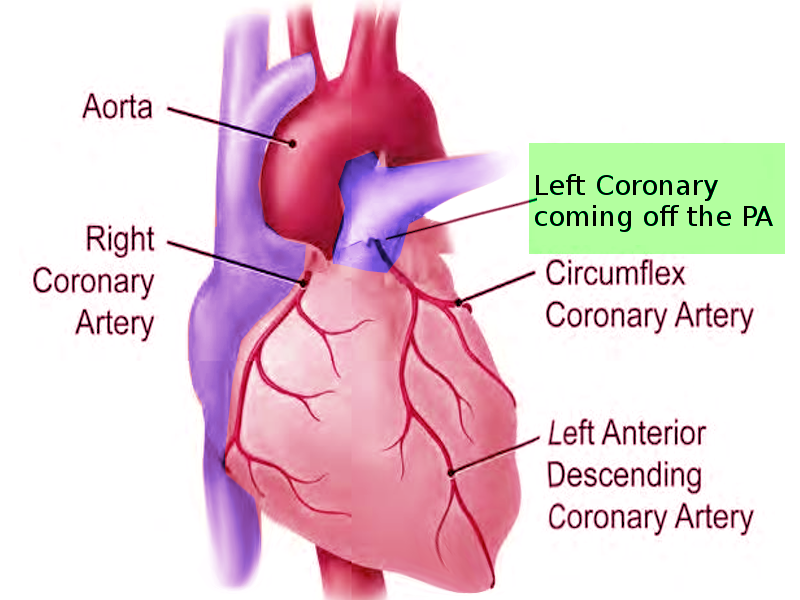
The left main coronary artery (LCA) classically arises from the ascending aorta just above the left cusp of the aortic valve. However, as with many of the coronary arteries, anomalies of the origin of the LCA have been well described. We will focus on two main anomalies: anomalous left main coronary artery with its origin from the pulmonary artery (ALCAPA) and anomalous left coronary artery origin from the right coronary sinus (ALCA-R). An LCA origin from the right coronary artery (RCA) has also been described.
Structure and Function
The LCA typically arises just distal to the left cusp of the aortic valve to supply blood to the left portion of the heart. After its take-off from the aorta, the LCA splits into the left circumflex (LCx) artery and the left anterior descending (LAD) artery. The LCx supplies the lateral portion of the left atrium and ventricle, while the LAD supplies the anterior portion of the left ventricle and the anterior 2/3 of the interventricular septum.
Physiologic Variants
ALCAPA
An anomalous left main coronary artery with its origin from the pulmonary artery (ALCAPA) is one of the few clinically significant coronary anomalies. While most congenital coronary artery abnormalities involve various origins of the vessels from the aorta and are usually benign, ALCAPA can be fatal either during infancy or result in sudden death in adulthood. The incidence of ALCAPA is between 1 in 30,000 and 1 in 300,000 infants and accounts for 0.25 to 0.5% of congenital cardiac defects. Albeit rare, it is the most common cause of myocardial infarction in pediatric patients.[1][2] Collaterals originating from the right coronary artery perfuse the left coronary artery (LCA), which subsequently drain into the pulmonary artery (PA). Less commonly, the defect involves the right, rather than the main pulmonary artery. Rarely the defect includes the circumflex, right coronary, or both coronary arteries. Perfusion of the anomalous left coronary is maintained by the elevated pulmonary artery pressures after birth. Without clinical appreciation and correction, this major defect is fatal
Approximately 15% of patients will have an associated anomaly. The literature describes ALCAPA in combination with ventricular septal defects (VSD), patent ductus arteriosus (PDA), tetralogy of Fallot (TOF), pulmonary valvular stenosis, coarctation of the great vessels, and one case of hypoplastic left heart syndrome.[1] Should the presence of one of these defects increase pulmonary artery pressure, there is a resultant decline in the degree of myocardial ischemia.
ALCA-R
When a coronary artery arises from the opposite sinus of Valsalva, it is the anomalous aortic origin of a coronary artery (AAOCA). The anomalous left coronary artery origin from the right coronary sinus (ALCA-R) involves the aberrant origin of the LCA from the right coronary sinus, which is the opposite of the classically described anatomy. After this takeoff, there is a large amount of variability in the path of the anomalous vessel. Three of these pathways are generally benign: The ALCA-R may track anterior to the RV outflow tract, retro-aortic, or directly into the myocardium through the conal septum. The final reported pathway is a significant cause of sudden cardiac death (SCD). This pathway is known as intramural, wherein the anomalous vessel travels in the wall of the aorta just above the commissure. A study of 17 cases of SCD due to an anomalous coronary artery arising from the incorrect ostium found that 10 cases were ARCA-L and 7 were ARCA-R. All cases of SCD due to an AAOCA resulted from an intramural (AKA intra-arterial) path of the anomalous vessel.[3][4][5]
LCA arising from the RCA
A third anomaly has been reported wherein the LCA arises from the main stem of the RCA. Unfortunately, this anomaly has not been as well described in the literature, and the overall impact of this anomaly is not well elucidated at this time. Further research could identify the sequelae of such a condition and outline its’ best management.
Surgical Considerations
ALCAPA
Without surgical correction, this anomaly is fatal. Simple ligation of the anomalous LCA was the first successful operation described for ALCAPA. Ligation of the LMCA from the PA excluded the left-to-right shunt, thus allowing the collaterals from the RCA to perfuse the ventricle. This procedure has since fallen out of favor due to the significant risk of sudden death. Additional surgical corrections include bypass, reimplantation, and in situ conduit procedures, with the ultimate goal of developing a dual coronary artery system. Bypass grafting has used the left subclavian artery, internal mammary artery, and saphenous vein graft, but the results have been disappointing. Ultimately, reimplantation, if possible, is the ideal technique. This approach depends on the left coronary artery to be sufficient length for mobilization to the left sinus of Valsalva. If arterial mobilization is an issue, an ostium can be created in another part of the aorta for reimplantation. Takeuchi fashioned a conduit to direct flow from the aorta to the anomalous artery by creating an aortopulmonary window and intrapulmonary baffle with a flap of the pulmonary artery.[6] Dehiscence of the baffle–a cited complication–results in the redevelopment of the left-to-right shunt. The outcome depends on the degree of irreversible ventricular dysfunction. However, neither severe ventricular dysfunction nor mitral insufficiency is a contraindication to revascularization, as significant recovery is the norm. Management of mitral insufficiency remains controversial. While the regurgitation resolves in most cases with revascularization, the extremely rare case of substantial myocardial ischemia with infarcted papillary muscles exists. In this case, valvular repair has shown good results.[7][8]
ALCA-R
Patients with an intraarterial course of the ALCA-R are typically managed surgically with a modified unroofing procedure. This procedure removes the intra-arterial course of the vessel and prevents ischemia. The morbidity associated with the procedure is low, but peri-operative ischemia has been reported on multiple occasions. Another study found that exercise limitation may show equal benefit to surgical modification. Surgical management should merit consideration in extreme cases of myocardial ischemia found on a cardiopulmonary exercise test.[9][10]
Clinical Significance
ALPCAPA
Symptoms usually occur within the first two months of life–the time the ductus arteriosus closes, as the clinical effect of ALCAPA is dependent on the pressure difference between the systemic and pulmonary circulatory systems. Decreased resistance in the pulmonary vasculature results in a left-to-right shunt from the right coronary artery to the PA–coronary steal phenomenon. The elevated pulmonary vascular resistance in utero results in systemic pulmonary arterial pressures. Consequently, the perfusion through the anomalous LCA is sufficient, albeit desaturated. Thus, symptoms rarely present early during infancy. Antegrade flow through the LCA decreases with the decline in PA pressure and pulmonary vascular resistance, and becomes more dependent on the collateral network between the right and left coronary arteries. The maturity of this collateral network will determine the clinical course after the ductal closure, as it ultimately determines the myocardial viability. A child with a well-developed collateral network may survive past infancy, albeit with progressive LV dysfunction. The lack of adequate collateral vessels from the RCA causes myocardial ischemia and ventricular dysfunction from insufficient perfusion, with eventual myocardial infarct, aneurysmal changes, and ultimately death. Early ischemic changes usually appear within the subendocardium with a patchy distribution.
The typical clinical symptoms appreciated at 4 to 6 weeks include poor feeding, tachypnea, failure to thrive, and pallor. With an enlarged cardiac silhouette seen on chest X-ray, children commonly receive a diagnosis of heart failure due to a dilated cardiomyopathy. Clinicians have confused a syndrome of severe pallor, diaphoresis, and irritability as infantile colic, in which the term "anginal equivalent" was coined.[2] It is pertinent to suspect an anomalous coronary defect in infants presenting with heart failure and dilated cardiomyopathy. In older children, due to mature collateral networks or associated defects, maintaining elevated pulmonary vascular resistance, complaints include angina, dyspnea, and fatigue. The defect may result in arrhythmias or even sudden death.
Physical exam findings include tachypnea, tachycardia, a gallop rhythm, cardiomegaly, hepatomegaly, and commonly a systolic ejection murmur due to mitral regurgitation. The murmur is a result of either papillary muscle dysfunction or ventricular remodeling. The enlarged cardiac silhouette on chest X-ray is nonspecific; however, pulmonary congestion is also common in infants. Lateral or anterolateral infarction with Q waves and elevated ST segments on electrocardiogram (ECG) are classic findings in infants. In older children and adults, the Q waves are less prominent; however, hypertrophy of the posterobasal left ventricular wall may be appreciated.
Diagnostic studies include echocardiogram and cardiac catheterization. A dilated, poorly contracting left ventricle is common on echocardiography, with mitral regurgitation due to a poorly mobile posterior leaflet. It is possible to visualize the coronary ostia on color-flow Dopper echocardiography; however, coronary angiography remains the best diagnostic tool and is necessary if color-flow Doppler does not identify both ostia with certainty. While the anomalous LMCA may originate anywhere along the main PA or one of the branches, it most commonly originates from the rightward posterior sinus. Coronary arteriograms will also show the collaterals originating from the RCA, late filling of the PA, and the left-to-right shunt. The cardiac catheterization will also document elevated LV end-diastolic, left atrial, and pulmonary artery pressures. Pulmonary angiography may be an option if doubt remains about the diagnosis, or the origin of the LCA ostium is uncertain. Magnetic resonance angiography (MRA) has been reported to be useful in defining the proximal course of the vessels.[11]
ALCA-R
The major clinical significance of the ALCA-R is the risk of myocardial ischemia and sudden cardiac death in the intraarterial course of the anomalous course. The ischemia is the result of compression of the anomalous vessel from surrounding tissue, an issue that is most significant during systole and exercise. The remaining three subtypes of the ALCA-R are only rarely associated with myocardial ischemia.
Unfortunately, the diagnosis of this condition is difficult. EKG typically shows no abnormalities, and the symptoms mimic those of angina. Patients in which there is suspicion of ALCA-R should undergo an evaluation with an MRA or CT angiogram followed by an intravascular ultrasound to determine the subtype and extent of compression of the anomalous vessels. The diagnosis is, unfortunately, most commonly made following an episode of SCD.[3][4]