Continuing Education Activity
Aniridia is defined as a partial or complete absence of the iris. It may be associated with various systemic disorders and multiple ocular morbidities. This activity reviews the evaluation and treatment of aniridia and highlights the role of the interprofessional team in evaluating and treating patients with this condition.
Objectives:
- Describe the etiology, epidemiology, and pathophysiology of aniridia.
- Explain the histopathology and common physical exam findings associated with aniridia.
- Review the management options of aniridia.
- Review the importance of improving care coordination amongst interprofessional team members to improve outcomes for patients affected by aniridia.
Introduction
Aniridia is defined as a partial or complete absence of the iris. Aniridia can be either congenital or acquired. Congenital aniridia is a rare disease that affects both eyes. It is autosomal dominant in the majority. Most of the other cases of congenital aniridia are sporadic. Sporadic aniridia may correlate with WAGR syndrome (Wilm tumor, aniridia, genitourinary anomalies, and mental retardation). A minority of congenital aniridia may be transmitted autosomal recessively called the Gillespie syndrome (aniridia, cerebellar ataxia, and mental retardation).
Research has noted a mutation of the PAX6 gene at chromosome 11p13 in congenital aniridia.
Acquired aniridia occurs after trauma or ocular surgery.
Etiology
The exact cause of congenital aniridia is unknown. However, the paired box gene (PAX6) gene at chromosome 11p13 is a crucial factor in pathogenesis. This gene is involved in the development of essential organs, including the eye, pancreas, brain, and spinal cord during embryonic development.
The various hypothesis regarding the etiopathogenesis of aniridia include:
- Aniridia may be a subtype of iris coloboma.
- Neuroectodermal theory - Aniridia develops from an anomaly of neuroectodermal development - this theory has support from the association of aniridia with neuroectodermal defects like the absence of iris muscles and foveal hypoplasia.
- Mesodermal theory suggests that there is an abnormality of the development of mesodermal tissues like optic nerve head hypoplasia; this does not explain foveal hypoplasia which commonly accompanies aniridia.
Classification of congenital aniridia:
Autosomal dominant congenital aniridia- It consists of at least two-thirds cases of congenital aniridia and may be present in up to 85% cases with aniridia. This variant is autosomal dominant and the most common form of aniridia; it has complete penetrance but variable expressivity. Thus family members may have different severity of aniridia, ocular involvement, and visual acuity. Some cases may have subtle iris hypoplasia or iris coloboma.
Sporadic congenital aniridia- There is a de novo mutation of the PAX6 gene and may consist of 13% to 33% of cases with aniridia. An important fact is that this variant of aniridia correlates with nephroblastoma (Wilms tumor) as a part of WAGR syndrome (Wilms tumor, aniridia, genitourinary anomalies, and mental retardation), also known as Miller syndrome. The genitourinary anomalies include undescended testes in males and streak gonads (nonfunctional ovaries) and/or bicornuate uterus in females. Intellectual disability includes difficulty in learning and processing information. There might be other manifestations, including attention deficit hyperactivity disorder, obsessive-compulsive disorder, depression, and anxiety. There may be craniofacial dysmorphism.
WAGR syndrome occurs as the chromosomal location of the PAX6 gene at 11p13 is very close to WT1 (Wilms tumor) gene. There is a partial deletion of the short arm of chromosome 11 involving both PAX6 and WT1 in this syndrome. Patients with WAGR syndrome have around 45 to 60% chance of developing nephroblastoma. Approximately 25% to 33% of patients with sporadic congenital aniridia may develop nephroblastoma before 3 years of age.[1]
Autosomal recessive congenital aniridia- It consists of 1% to 3% of all congenital aniridia cases and is the least common variant of congenital aniridia. It has associations with cerebellar ataxia and mental retardation (Gillespie syndrome).
Epidemiology
Aniridia is noted in around 1.8 of 100,000 births.[2] Males and females are equally affected.
Pathophysiology
Various genes implicated in the pathogenesis of congenital aniridia include:
Aniridia 1 or AN1 (OMIM # 106210)- This results from heterozygous mutation of the PAX6 gene at chromosome 11p13. It is inherited autosomal dominantly, and also includes late-onset corneal dystrophy and congenital cataracts. Other associations include Peters anomaly, reduced smell sensation, hypoplasia of corpus callosum and/or anterior commissure, and the absence of the pineal gland.
Aniridia 2 (AN2) (OMIM # 617141, autosomal dominant) results from the mutation of a cis-regulatory element of the PAX6 gene. This regulatory region is at the intron of the adjacent ELP4 gene (11p13).
Aniridia 3 (AN3) (OMIM # 617142, autosomal dominant) is due to a mutation in the TRIM44 gene (11p13).
Gillespie syndrome (OMIM # 206700) occurs due to a mutation of the ITPR1 gene at chromosome 3p26.1.
Research has suggested a probable linkage of autosomal dominant aniridia and acid phosphatase 1 (ACP1) at chromosome 2p.[3]
Histopathology
The histopathology shows arrest of neuroectodermal development, which includes absent iris muscles with hypoplasia of iris. There may be dysgenesis of the Schlemm's canal and trabecular meshwork. Hypoplasia of the ciliary body is also noted but is less severe than iris-hypoplasia. Large iris vessels may be present over the anterior surface of the hypoplastic iris. The anterior chamber angle may be normal. There may be incomplete cleavage of the angle or anomalous development of the angle.[4]
History and Physical
The patients usually complain of photophobia related to the severity of aniridia and poor vision (due to many reasons, including foveal hypoplasia and optic nerve head hypoplasia) since early childhood. Parents may bring the child for the abnormal appearance of the eyes.
Nystagmus and strabismus are common due to poor vision since childhood.
Other ocular findings include:
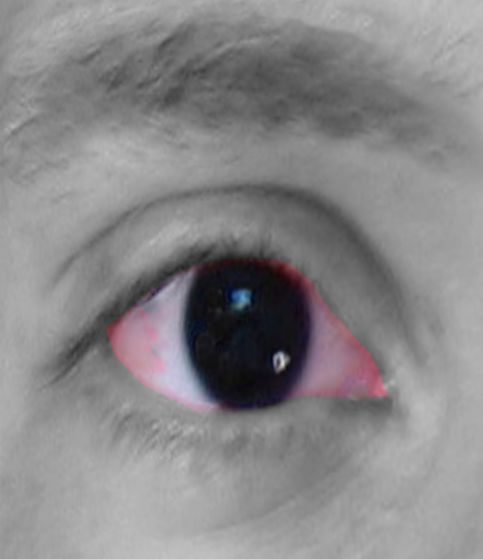
- Aniridia- The iris may be partially absent (detectable with retroillumination), or there may be an apparent complete absence of iris on external observation. However, most cases have small hypoplastic iris tissue visible on gonioscopy. All cases have some hypoplastic iris tissue on histopathological examination.[1] Thus, the term aniridia is a misnomer, and 'iridemia' may be a better term to describe the disease. There may be iris coloboma, corectopia (eccentric pupil), and loss of crypts of iris.
- Bilateral ptosis
- Meibomian gland dysfunction
- Aniridia associated keratopathy (AAK)- There is limbal stem cell deficiency, corneal pannus, corneal epithelial irregularity, and conjunctivalization of the cornea. Corneal opacity, corneal scarring (subepithelial fibrosis), and vascularization are seen peripherally and may encroach the central cornea. Corneal portends a bad prognosis after penetrating keratoplasty and predisposes to graft rejection and failure. There is dry eye and tear film instability. The ocular surface may be very poor and unstable predisposing to epithelial defect and/or infective keratitis. A classification of AAK into three stages (mild, moderate, and severe limbal insufficiency) has been suggested.[5][6]
- Microcornea
- Glaucoma - The corneal thickness is usually high, which might influence the measurement of the intraocular pressure. In the early years of life, the angle of the anterior chamber is generally open on gonioscopy, and congenital glaucoma/buphthalmos is rare in aniridia. However, within 2nd-decade angle closure due to peripheral anterior synechia is noted, which may lead to an increase in the intraocular pressure.
- Persistent pupillary membrane
- Rudimentary iris stump on gonioscopy - Gonioscopy may show iris fibers which lie at the space between the rudimentary small hypoplastic iris and the trabecular meshwork. These fibers contract and pull the rudimentary iris forward thereby causing synechial angle-closure glaucoma
- Superior subluxation of the lens (clear or cataractous), ectopia lentis (noted in up to 56% of cases),[1] dislocation of the lens
- Cataract or opacities of the crystalline lens or its capsule may be present in 50% to 85% of patients.[1]
- Microspherophakia
- Optic nerve head hypoplasia is present in 75% of cases in a series of 12 patients with aniridia.[7] A reduced disc diameter may confirm the hypoplasia: width of the retinal arteriole at the disc margin of 6.1 to less than 14.6 (normal value 14.6 ± 2.4)[2]
- Foveal hypoplasia - Foveal hypoplasia, characterized by the absence of a defined foveal avascular zone, the absence of pigmentation at the fovea, and blood vessels may cross through the area where fovea is presumed to be present. Foveal depression is absent, and inner retinal layers are present all over the macular region. In a normal fovea, inner retinal layers are absent, and there is a depression corresponding to the foveola.
- Fundal coloboma
- Aniridic fibrosis syndrome[8]- 'A progressive anterior fibrosis syndrome in patients with postsurgical congenital aniridia' has been described. Usually, there is a history of multiple surgeries, including cataract surgery, glaucoma surgery, and keratoplasty. This condition is characterized by retrocorneal and retrolenticular membranes arising from the rudimentary iris, which may involve ciliary processes and anterior retina also. This progressive membrane results in anterior displacement of the intraocular lens and corneal decompensation.
Other associations of aniridia include:
- Sensory deficiency including
- Abnormal olfaction
- Hearing loss[9]
- AGR triad (aniridia, genitourinary abnormalities, mental retardation)
- Absent or hypoplastic patella (OMIM 106220, aniridia and absent patella) in family
Evaluation
The fundus fluorescein angiogram reveals the lack of a well-defined foveal avascular zone.
Optical coherence tomography in foveal hypoplasia shows the absence of foveal depression and the presence of the inner retinal layers.
In sporadic congenital aniridia, Wilms tumor must be ruled out by regular ultrasonogram of the abdomen. Also, genetic evaluation for PAX6 and WT1 gene is possible.
Ophthalmic examination of the family members is of utmost importance.
Treatment / Management
In early childhood, the management includes optimal refractive correction, amblyopia therapy, and correction of squint.
Painted contact lenses may reduce photophobia, improve cosmesis, and improve vision. Tinted contact lenses may also be an option. Such contact lenses also reduce nystagmus. Photochromatic or tinted glasses may be helpful.
The food and drug administration has approved an artificial iris implant. It may improve the disabling glare, light sensitivity during day or night, improve cosmesis, and vision-related quality of life. However, there is a risk of development or worsening of glaucoma and limbal stem cell deficiency. An artificial iris may be used to treat
- Aniridia (congenital or traumatic)
- Iris defects due to various causes, including albinism, or after excision of iris mass.
Glaucoma- The medications usually fail to control glaucoma, and surgical intervention is needed. Glaucoma presenting before 1 year of age, is very difficult to treat. Surgery is often necessary for glaucoma by 20 years of age in patients with congenital aniridia. The options for surgical therapy include:
- Goniotomy
- Trabeculotomy
- Trabeculectomy with mitomycin C
- Combined trabeculectomy and trabeculotomy
- Glaucoma drainage devices may be implanted as a primary modality in aniridic glaucoma.[10]
The success of surgical intervention may not be significant. In a study, success rates for various interventions were:
- 9% for trabeculectomy
- 25% for cyclocryotherapy; however side effects of phthisis and cataract was noted
- 83% for Molteno implant[11]
Okada and colleagues noted that 'mean good intraocular pressure control period after the filtering surgery was 14.6 months'.[12] The success rate of trabeculectomy is better in older patients, and Nelson and colleagues reported successful trabeculectomies in 11 of 14 cases with the minimum follow-up of 1 year.[1] Ultimately, if other modalities fail to control intraocular pressure, cycloablative therapies may be needed, which include diode laser cyclophotocoagulation and cyclocryotherapy. Prophylactic goniotomy may prevent the development of glaucoma in congenital aniridia.[13]
The cataract in aniridia may need extraction. Surgery is difficult due to zonular weakness and fragility of the lens capsule. The eye may be left aphakic, and postoperatively contact lens or aphakic glasses may be prescribed. A special intraocular lens (IOL) for aniridia is available, which has a peripheral colored region like iris of different pigmentation. Tinted IOLs are also available, which can reduce photophobia. In extreme subluxation, scleral-fixated IOL may be necessary. Surgical trauma may increase the chances of glaucoma, corneal decompensation, and limbal stem cell deficiency.
Aniridia-associated keratopathy:
Phase 0 (subclinical limbal stem cell deficiency and phase 1 (slight limbal stem cell deficiency, less than 2 corneal epithelial erosion or ulcer in last 6months, corneal pannus less than 1mm from limbus, mild watering and photophobia, and fluorescein staining abnormality)[14] The dry eye and keratopathy need frequent artificial tear drops (free of preservatives). Antiglaucoma drops with preservatives may worsen the corneal surface.
Phase 2 (Moderate limbal insufficiency, constant redness, watering, and photophobia, more than three corneal erosions or ulcers in the last six months, constant tear film instability, vascular pannus)[14] - amniotic membrane transplant or autologous serum is used.
Phase 3 (severe limba insufficiency, corneal vascularization, loss of vision)- Autologous transplant of limbal epithelial cells is not possible due to bilateral involvement. Management options include:
- 2 stage surgery involving initial replenishment of the limbal stem cells followed by surgery for visual rehabilitation
- Options for replenishing limbal stem cells include:
- Keratolimbal allografts
- Cultured limbal stem cell transplant
- Cultured oral mucosal epithelial transplantation [15]
- Options for visual rehabilitation include
- Deep anterior lamellar keratoplasty
- Penetrating keratoplasty
- Simultaneous surgeries for both replenishment of the limbal stem cells and restoration of media clarity by keratoplasty have also been successful.[16]
- Boston keratoprosthesis may be helpful in the visual rehabilitation of patients with severe aniridia-associated keratopathy.[17]
However, the survival of corneal graft is often poor due to multiple causes, including corneal vascularization, limbal stem cell deficiency, severe dry eye, and glaucoma.
The associated squint and ptosis may need correction.
Aniridic fibrosis syndrome needs early surgical intervention to remove the membranes.
Systemic diseases need a collaborative approach of specialists of different subspecialties.
Differential Diagnosis
- Congenital Cataract
- Congenital Nystagmus
- Ectopia Lentis
- Iris Coloboma
- Juvenile Glaucoma
Prognosis
Visual prognosis is usually poor. Up to 86% of patients with aniridia may have a visual acuity of 20/100 or worse in the better eye. The cause of visual decline include:
- Foveal hypoplasia
- Optic nerve head hypoplasia
- Glaucoma
- Cataract
- Keratopathy
However, members of some families with familial aniridia may have good vision of at least 20/30 in up to 61% of cases.[18]
Complications
Aniridia may be complicated by:
- Glaucoma - Around 50% to 75% of patients with aniridia develop glaucoma, which, however, is usually not seen at birth. It usually occurs after 10 to 20 years of age likely due to blockage of the trabecular meshwork by the contracting iris strands of the rudimentary iris tissue or maldeveloped angle of the anterior chamber (goniodysgenesis). Glaucoma is the predominant cause of blindness in aniridia. The treatment of glaucoma is complicated, and the glaucoma is often resistant to both medical and surgical treatment. The prognosis of glaucoma associated with aniridia is guarded.
- Aniridic fibrosis syndrome
- Painful or painless blind eye
- Systemic associations including Wilms tumor
Pearls and Other Issues
All patients with sporadic congenital aniridia should be screened regularly with abdominal ultrasonography for Wilms tumor unless a genetic evaluation rules out mutation or deletion of WT1. A proposed protocol of ultrasound screening is
- Up to 5 years of age: every 3 months
- 5 to 10 years of age: every 6 months
- 10 to 16 years of age: every 12 months
Enhancing Healthcare Team Outcomes
Diagnosing aniridia and properly managing it requires a team effort. There should be proper coordination between primary care providers, pediatric ophthalmologists, geneticist, and ophthalmology nurses to reach a conclusion regarding the management plan and to counsel the parents accordingly. The nurses care for patients perioperatively, provide family education, and update the team on the patient's condition. The prognosis for most patients as far as visual acuity is concerned is poor.[19][Level V] Interprofessional collaboration is crucial for better outcomes and reduce mortality of patients with aniridia and systemic diseases. Specifically, sporadic congenital aniridia cases must be screened regularly for Wilms tumor.