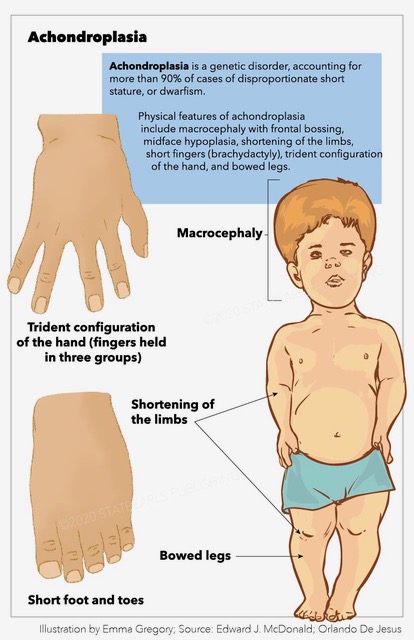
[1]
Vajo Z, Francomano CA, Wilkin DJ. The molecular and genetic basis of fibroblast growth factor receptor 3 disorders: the achondroplasia family of skeletal dysplasias, Muenke craniosynostosis, and Crouzon syndrome with acanthosis nigricans. Endocrine reviews. 2000 Feb:21(1):23-39
[PubMed PMID: 10696568]
[2]
Wang X, Ramström O, Yan M. Glyconanomaterials: synthesis, characterization, and ligand presentation. Advanced materials (Deerfield Beach, Fla.). 2010 May 4:22(17):1946-53. doi: 10.1002/adma.200903908. Epub
[PubMed PMID: 20301131]
[3]
Bellus GA, Hefferon TW, Ortiz de Luna RI, Hecht JT, Horton WA, Machado M, Kaitila I, McIntosh I, Francomano CA. Achondroplasia is defined by recurrent G380R mutations of FGFR3. American journal of human genetics. 1995 Feb:56(2):368-73
[PubMed PMID: 7847369]
[4]
Hecht JT, Francomano CA, Horton WA, Annegers JF. Mortality in achondroplasia. American journal of human genetics. 1987 Sep:41(3):454-64
[PubMed PMID: 3631079]
[5]
Horton WA. Recent milestones in achondroplasia research. American journal of medical genetics. Part A. 2006 Jan 15:140(2):166-9
[PubMed PMID: 16353253]
[7]
Waller DK, Correa A, Vo TM, Wang Y, Hobbs C, Langlois PH, Pearson K, Romitti PA, Shaw GM, Hecht JT. The population-based prevalence of achondroplasia and thanatophoric dysplasia in selected regions of the US. American journal of medical genetics. Part A. 2008 Sep 15:146A(18):2385-9. doi: 10.1002/ajmg.a.32485. Epub
[PubMed PMID: 18698630]
[8]
Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, Legare JM. Achondroplasia. GeneReviews(®). 1993:():
[PubMed PMID: 20301331]
[9]
Oberklaid F, Danks DM, Jensen F, Stace L, Rosshandler S. Achondroplasia and hypochondroplasia. Comments on frequency, mutation rate, and radiological features in skull and spine. Journal of medical genetics. 1979 Apr:16(2):140-6
[PubMed PMID: 458831]
Level 3 (low-level) evidence
[10]
Coi A, Santoro M, Garne E, Pierini A, Addor MC, Alessandri JL, Bergman JEH, Bianchi F, Boban L, Braz P, Cavero-Carbonell C, Gatt M, Haeusler M, Klungsøyr K, Kurinczuk JJ, Lanzoni M, Lelong N, Luyt K, Mokoroa O, Mullaney C, Nelen V, Neville AJ, O'Mahony MT, Perthus I, Rankin J, Rissmann A, Rouget F, Schaub B, Tucker D, Wellesley D, Wisniewska K, Zymak-Zakutnia N, Barišić I. Epidemiology of achondroplasia: A population-based study in Europe. American journal of medical genetics. Part A. 2019 Sep:179(9):1791-1798. doi: 10.1002/ajmg.a.61289. Epub 2019 Jul 11
[PubMed PMID: 31294928]
[11]
Kovac JR, Addai J, Smith RP, Coward RM, Lamb DJ, Lipshultz LI. The effects of advanced paternal age on fertility. Asian journal of andrology. 2013 Nov:15(6):723-8. doi: 10.1038/aja.2013.92. Epub 2013 Aug 5
[PubMed PMID: 23912310]
[12]
Wyrobek AJ, Eskenazi B, Young S, Arnheim N, Tiemann-Boege I, Jabs EW, Glaser RL, Pearson FS, Evenson D. Advancing age has differential effects on DNA damage, chromatin integrity, gene mutations, and aneuploidies in sperm. Proceedings of the National Academy of Sciences of the United States of America. 2006 Jun 20:103(25):9601-6
[PubMed PMID: 16766665]
[13]
Shirley ED, Ain MC. Achondroplasia: manifestations and treatment. The Journal of the American Academy of Orthopaedic Surgeons. 2009 Apr:17(4):231-41
[PubMed PMID: 19307672]
[14]
Laederich MB, Horton WA. Achondroplasia: pathogenesis and implications for future treatment. Current opinion in pediatrics. 2010 Aug:22(4):516-23. doi: 10.1097/MOP.0b013e32833b7a69. Epub
[PubMed PMID: 20601886]
Level 3 (low-level) evidence
[15]
Horton WA, Rotter JI, Rimoin DL, Scott CI, Hall JG. Standard growth curves for achondroplasia. The Journal of pediatrics. 1978 Sep:93(3):435-8
[PubMed PMID: 690757]
[16]
Wigg K, Tofts L, Benson S, Porter M. The neuropsychological function of children with achondroplasia. American journal of medical genetics. Part A. 2016 Nov:170(11):2882-2888. doi: 10.1002/ajmg.a.37779. Epub 2016 Sep 8
[PubMed PMID: 27605460]
[17]
Kitoh H, Kitakoji T, Kurita K, Katoh M, Takamine Y. Deformities of the elbow in achondroplasia. The Journal of bone and joint surgery. British volume. 2002 Jul:84(5):680-3
[PubMed PMID: 12188484]
[18]
Kopits SE. Genetics clinics of The Johns Hopkins Hospital. Surgical intervention in achondroplasia. Correction of bowleg deformity in achondroplasia. The Johns Hopkins medical journal. 1980 May:146(5):206-9
[PubMed PMID: 7382244]
[19]
Inan M, Thacker M, Church C, Miller F, Mackenzie WG, Conklin D. Dynamic lower extremity alignment in children with achondroplasia. Journal of pediatric orthopedics. 2006 Jul-Aug:26(4):526-9
[PubMed PMID: 16791073]
[20]
Kopits SE. Thoracolumbar kyphosis and lumbosacral hyperlordosis in achondroplastic children. Basic life sciences. 1988:48():241-55
[PubMed PMID: 3240259]
[21]
Margalit A, McKean G, Lawing C, Galey S, Ain MC. Walking Out of the Curve: Thoracolumbar Kyphosis in Achondroplasia. Journal of pediatric orthopedics. 2018 Nov/Dec:38(10):491-497. doi: 10.1097/BPO.0000000000000862. Epub
[PubMed PMID: 27636912]
[22]
Steinbok P, Hall J, Flodmark O. Hydrocephalus in achondroplasia: the possible role of intracranial venous hypertension. Journal of neurosurgery. 1989 Jul:71(1):42-8
[PubMed PMID: 2786928]
[23]
Berkowitz RG, Grundfast KM, Scott C, Saal H, Stern H, Rosenbaum K. Middle ear disease in childhood achondroplasia. Ear, nose, & throat journal. 1991 May:70(5):305-8
[PubMed PMID: 1914954]
[24]
Ireland PJ, Donaghey S, McGill J, Zankl A, Ware RS, Pacey V, Ault J, Savarirayan R, Sillence D, Thompson E, Townshend S, Johnston LM. Development in children with achondroplasia: a prospective clinical cohort study. Developmental medicine and child neurology. 2012 Jun:54(6):532-7. doi: 10.1111/j.1469-8749.2012.04234.x. Epub 2012 Mar 12
[PubMed PMID: 22409389]
[25]
Hoover-Fong JE, McGready J, Schulze KJ, Barnes H, Scott CI. Weight for age charts for children with achondroplasia. American journal of medical genetics. Part A. 2007 Oct 1:143A(19):2227-35
[PubMed PMID: 17764078]
[26]
Jiang ZH. [Conray ventriculography in the diagnosis of tumors in the posterior cranial fossa (author's transl)]. Zhonghua fang she xue za zhi Chinese journal of radiology. 1979:13(2):84-7
[PubMed PMID: 162380]
[27]
Bagley CA, Pindrik JA, Bookland MJ, Camara-Quintana JQ, Carson BS. Cervicomedullary decompression for foramen magnum stenosis in achondroplasia. Journal of neurosurgery. 2006 Mar:104(3 Suppl):166-72
[PubMed PMID: 16572633]
[28]
Engberts AC, Jacobs WC, Castelijns SJ, Castelein RM, Vleggeert-Lankamp CL. The prevalence of thoracolumbar kyphosis in achondroplasia: a systematic review. Journal of children's orthopaedics. 2012 Mar:6(1):69-73. doi: 10.1007/s11832-011-0378-7. Epub 2011 Dec 3
[PubMed PMID: 22442656]
Level 1 (high-level) evidence
[29]
Abousamra O, Shah SA, Heydemann JA, Kreitz TM, Rogers KJ, Ditro C, Mackenzie WG. Sagittal Spinopelvic Parameters in Children With Achondroplasia. Spine deformity. 2019 Jan:7(1):163-170. doi: 10.1016/j.jspd.2018.06.001. Epub
[PubMed PMID: 30587311]
[30]
Ain MC, Shirley ED, Pirouzmanesh A, Hariri A, Carson BS. Postlaminectomy kyphosis in the skeletally immature achondroplast. Spine. 2006 Jan 15:31(2):197-201
[PubMed PMID: 16418640]
[31]
Monticelli G, Spinelli R. Leg lengthening by closed metaphyseal corticotomy. Italian journal of orthopaedics and traumatology. 1983 Jun:9(2):139-50
[PubMed PMID: 6654651]
[32]
Semler O, Rehberg M, Mehdiani N, Jackels M, Hoyer-Kuhn H. Current and Emerging Therapeutic Options for the Management of Rare Skeletal Diseases. Paediatric drugs. 2019 Apr:21(2):95-106. doi: 10.1007/s40272-019-00330-0. Epub
[PubMed PMID: 30941653]
[33]
Breinholt VM, Rasmussen CE, Mygind PH, Kjelgaard-Hansen M, Faltinger F, Bernhard A, Zettler J, Hersel U. TransCon CNP, a Sustained-Release C-Type Natriuretic Peptide Prodrug, a Potentially Safe and Efficacious New Therapeutic Modality for the Treatment of Comorbidities Associated with Fibroblast Growth Factor Receptor 3-Related Skeletal Dysplasias. The Journal of pharmacology and experimental therapeutics. 2019 Sep:370(3):459-471. doi: 10.1124/jpet.119.258251. Epub 2019 Jun 24
[PubMed PMID: 31235532]
[34]
Savarirayan R, Irving M, Bacino CA, Bostwick B, Charrow J, Cormier-Daire V, Le Quan Sang KH, Dickson P, Harmatz P, Phillips J, Owen N, Cherukuri A, Jayaram K, Jeha GS, Larimore K, Chan ML, Huntsman Labed A, Day J, Hoover-Fong J. C-Type Natriuretic Peptide Analogue Therapy in Children with Achondroplasia. The New England journal of medicine. 2019 Jul 4:381(1):25-35. doi: 10.1056/NEJMoa1813446. Epub 2019 Jun 18
[PubMed PMID: 31269546]
[35]
Krakow D, Rimoin DL. The skeletal dysplasias. Genetics in medicine : official journal of the American College of Medical Genetics. 2010 Jun:12(6):327-41. doi: 10.1097/GIM.0b013e3181daae9b. Epub
[PubMed PMID: 20556869]
[36]
Almeida MR, Campos-Xavier AB, Medeira A, Cordeiro I, Sousa AB, Lima M, Soares G, Rocha M, Saraiva J, Ramos L, Sousa S, Marcelino JP, Correia A, Santos HG. Clinical and molecular diagnosis of the skeletal dysplasias associated with mutations in the gene encoding Fibroblast Growth Factor Receptor 3 (FGFR3) in Portugal. Clinical genetics. 2009 Feb:75(2):150-6. doi: 10.1111/j.1399-0004.2008.01123.x. Epub
[PubMed PMID: 19215249]
[37]
Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, French T, Savarirayan R. Thanatophoric Dysplasia. GeneReviews(®). 1993:():
[PubMed PMID: 20301540]
[38]
Bellus GA, Spector EB, Speiser PW, Weaver CA, Garber AT, Bryke CR, Israel J, Rosengren SS, Webster MK, Donoghue DJ, Francomano CA. Distinct missense mutations of the FGFR3 lys650 codon modulate receptor kinase activation and the severity of the skeletal dysplasia phenotype. American journal of human genetics. 2000 Dec:67(6):1411-21
[PubMed PMID: 11055896]
[39]
Tavormina PL, Bellus GA, Webster MK, Bamshad MJ, Fraley AE, McIntosh I, Szabo J, Jiang W, Jabs EW, Wilcox WR, Wasmuth JJ, Donoghue DJ, Thompson LM, Francomano CA. A novel skeletal dysplasia with developmental delay and acanthosis nigricans is caused by a Lys650Met mutation in the fibroblast growth factor receptor 3 gene. American journal of human genetics. 1999 Mar:64(3):722-31
[PubMed PMID: 10053006]
[40]
Bellus GA, Bamshad MJ, Przylepa KA, Dorst J, Lee RR, Hurko O, Jabs EW, Curry CJ, Wilcox WR, Lachman RS, Rimoin DL, Francomano CA. Severe achondroplasia with developmental delay and acanthosis nigricans (SADDAN): phenotypic analysis of a new skeletal dysplasia caused by a Lys650Met mutation in fibroblast growth factor receptor 3. American journal of medical genetics. 1999 Jul 2:85(1):53-65
[PubMed PMID: 10377013]
[41]
Smid CJ, Modaff P, Alade A, Legare JM, Pauli RM. Acanthosis nigricans in achondroplasia. American journal of medical genetics. Part A. 2018 Dec:176(12):2630-2636. doi: 10.1002/ajmg.a.40506. Epub 2018 Oct 31
[PubMed PMID: 30380187]
[42]
Dai L, Xie L, Wang Y, Mao M, Li N, Zhu J, Kim C, Zhang Y. A novel COMP mutation in a pseudoachondroplasia family of Chinese origin. BMC medical genetics. 2011 May 21:12():72. doi: 10.1186/1471-2350-12-72. Epub 2011 May 21
[PubMed PMID: 21599986]
[43]
Ozcetin M, Arslan MT, Karapinar B. An achondroplasic case with foramen magnum stenosis, hydrocephaly, cortical atrophy, respiratory failure and sympathetic dysfunction. Iranian journal of pediatrics. 2012 Mar:22(1):121-4
[PubMed PMID: 23056871]
Level 3 (low-level) evidence