Continuing Education Activity
Right ventricular hypertrophy (RVH) is a pathologic increase in the right ventricular muscle mass in response to chronic pressure overload. Longtime lung disease is the most common cause. However, other cardiovascular conditions can also lead to RVH, such as congenital heart disease, interstitial lung disease, and chronic left heart failure. Affected individuals present with symptoms of pulmonary hypertension, including exertional chest pain and syncope, peripheral edema, dyspnea, and right upper quadrant pain. New symptoms can exacerbate chronic manifestations.
Electrocardiography and echocardiography are the mainstay diagnostic modalities. Besides treating the underlying cause, RVH is managed using diuretics, oxygen, and anticoagulants. The prognosis depends on the etiology and severity of pulmonary hypertension. Collaboration between healthcare professionals, including the primary care clinician and cardiologist, is essential to optimize patients' quality of life.
This activity for healthcare workers is designed to enhance learners' competence in evaluating and managing RVH. Participants improve their proficiency in distinguishing RVH from other cardiac conditions with similar presentations and formulate individualized treatment plans using evidence-based management strategies. Learners strengthen their diagnostic and management skills to collaborate effectively with an interprofessional team caring for patients with this condition.
Objectives:
Identify the clinical signs and symptoms indicative of right ventricular hypertrophy.
Screen high-risk patient populations, such as those with underlying cardiac conditions or chronic pulmonary diseases, for early detection of right ventricular hypertrophy.
Create an evaluation plan for a patient suspected to have right ventricular hypertrophy.
Implement effective care coordination strategies within the interprofessional team to formulate short- and long-term treatment strategies and enhance outcomes for patients with right ventricular hypertrophy.
Introduction
Right ventricular hypertrophy (RVH) is an abnormal enlargement or pathologic increase in the right ventricular muscle mass as a maladaptive response to chronic pressure overload. RVH most commonly arises from chronic, severe lung disease. The right ventricle is considerably smaller than the left in the normal heart, producing electrical forces largely obscured by the larger left ventricle.[1][2][3] By comparison, RVH leads to right axis deviation (RAD) and reversal of the normal R wave progression in the precordial leads. Pulmonary hypertension of any cause and tricuspid valve conditions, commonly regurgitation, often lead to right ventricular impairment.
Anatomy of the Right Ventricle
The right ventricle's complex geometry poses function assessment challenges. A thorough evaluation necessitates diverse imaging planes and acoustic windows due to the differences between the structures and functions of the right and left ventricles. These ventricles originate from separate embryonic origins. The left ventricle arises from the heart tube, whereas the right ventricle derives from the anterior heart field.
The right ventricle comprises inflow (sinus) and outflow (conus) regions, separated by a muscular ridge, the crista supraventricularis. The inflow region includes the tricuspid valve, chordae or papillary muscles, and right ventricular body. The right ventricle's free wall forms its boundaries, extending from the anterior and posterior aspects of the interventricular septum. The standard septal curvature forms a convexity toward the right ventricular cavity and imparts a crescent shape to the ventricle when cross-sectioned. The right ventricle's interior surface is heavily trabeculated. This feature, along with the moderator band and more apical insertion of the tricuspid valve's annulus, imparts key morphologic differences distinguishing the right from the left ventricle on echocardiography. The infundibulum is the right ventricle's smooth, funnel-shaped outflow portion ending at the pulmonic valve.
A standard RV free-wall thickness of 0.3 to 0.5 cm imparts a greater distensibility and larger cavity volumes in the right than the left ventricle due to lower end-diastolic filling pressures. Thus, the right ventricle's ejection fraction is typically 35% to 45%—less than the left ventricle's 55% to 65%—though both chambers generate identical stroke volumes.
Physiology of Right Ventricle
Preload, afterload, and intrinsic contractility abnormalities can cause ventricular dysfunction. Muscle fiber orientation variations in the right ventricle determine its symmetrical shortening in the longitudinal and radial planes. Longitudinal shortening contributes significantly more to ejection than in the left ventricle. The pronounced right ventricle shortening along the longitudinal axis facilitates systolic function measurement using straightforward techniques that avoid geometric assumptions or meticulous endocardial definition, addressing limitations in noninvasive assessment.
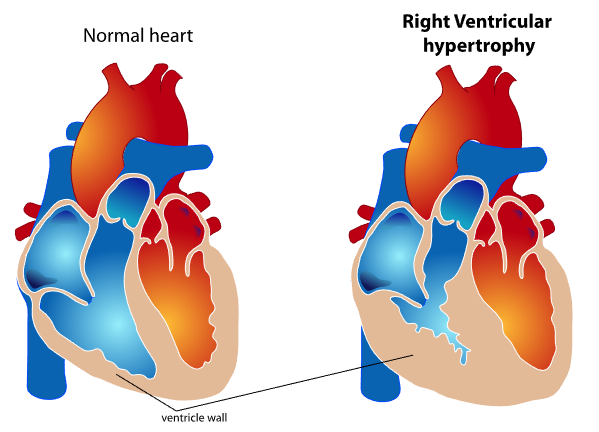
Key features distinguishing right from left ventricular physiology include a 2-layered muscle (instead of 3), resulting in longitudinal shortening, heightened sensitivity to pressure overload (as opposed to volume overload seen the left ventricular dysfunction), lower energy requirements for maintaining stroke volume, greater reliance (50%) on direct oxygen extraction from intraventricular blood rather than coronary blood flow, shorter action potential duration, and greater density of adrenergic and cholinergic receptors compared to the left ventricle. The predominantly longitudinal shortening leads to longitudinal motion instead of the wringing torsional motion observed in the left ventricle. Due to its thinner wall, the right ventricle is more susceptible to minor changes in pulmonary vascular resistance (PVR). Chronic pressure overload can induce RVH, ultimately progressing to right ventricular failure if left untreated.[4]
Etiology
RVH is a morphofunctional manifestation of an underlying disease process. The common causes of this condition are categorized as follows:
Right Ventricular Hypertrophy Due to Pulmonary Hypertension
Primary lung diseases are the most common causes of pulmonary hypertension leading to RVH. The World Health Organization (WHO) pulmonary hypertension classification system categorizes pulmonary hypertension into 5 groups based on etiology and pathophysiology.
- Group I: Pulmonary arterial hypertension (PAH)
- Group II: Pulmonary hypertension owing to left heart disease, such as mitral stenosis
- Group III: Pulmonary hypertension from lung disease, hypoxia, or both
- Group IV: Chronic thromboembolic pulmonary hypertension (CTEPH)
- Group V: Pulmonary hypertension with unclear mechanisms
Right Ventricular Hypertrophy From Congenital Heart Diseases
RVH is known to occur in various congenital heart diseases, including the following:
- Tetralogy of Fallot
- Isolated pulmonary stenosis
- Double-chambered right ventricle
- The systemic right ventricle in post-atrial switch heart
- Eisenmenger syndrome
Right Ventricular Hypertrophy From Intrinsic Myocardial Disease
Retrograde flow from a failing left ventricle can cause pulmonary hypertension and right ventricular dysfunction. Some conditions that may lead to this pathology include the following:
- Hypertrophic cardiomyopathy
- Cardiac fibrosis
- Athlete's heart
- Cardiac amyloidosis
- Essential hypertension
- Left heart failure
Right Ventricular Hypertrophy From Tricuspid Valve Disease
The tricuspid valvular condition most frequently causing RVH is tricuspid regurgitation. The valve does not close properly during systole, allowing blood to reflux into the right atrium. This regurgitant flow increases the right ventricle's volume load, requiring it to work harder to maintain sufficient cardiac output. Over time, the increased right ventricular workload and pressure overload lead to hypertrophy. The causes of tricuspid regurgitation include the following:
- Secondary tricuspid regurgitation mostly due to pulmonary or cardiac disease
- Primary tricuspid regurgitation arising from cardiac instrumentation, chest trauma, Ebstein anomaly, rheumatic fever, infective and marantic endocarditis, right ventricular ischemia or infarction, connective tissue disorder, carcinoid syndrome, and drug reactions [5][6][7][8]
Epidemiology
Large-scale studies to determine RVH prevalence are lacking. However, scattered research has investigated its occurrence across different disease states. For instance, a study found that RVH prevalence in 330 consecutive out-of-hospital hypertensive patients was 33.6%.[9] Another reported that extreme RVH prevalence in 2413 individuals with hypertrophic cardiomyopathy was 1.3%.[10] RVH prevalence in athletes varies based on the criteria used.
The condition's prevalence can also be inferred from pulmonary hypertension's epidemiology, which varies depending on the underlying cause. For instance, PAH (idiopathic and familial) has an estimated prevalence ranging from 10 to 52 cases per million adults worldwide. The exact prevalence of pulmonary hypertension from left heart failure is not well defined due to study design, pulmonary hypertension definition, and diagnostic modality variations. However. most echocardiography-based series suggest that left heart disease causes 70% of pulmonary hypertension cases.
Group 3 pulmonary hypertension appears to be more prevalent in older adults. A study of patients aged 65 and older with pulmonary hypertension revealed that group 3 pulmonary hypertension in this cohort occurred in 14%, while 28% had group 2 pulmonary hypertension, and 17% had mixed groups 2 and 3 pulmonary hypertension. CTEPH's prevalence is unknown but estimated to be between 1% and 5% among individuals who survive acute pulmonary embolism.
Pathophysiology
Comparing the pressure-volume loops of the adapting and failing right ventricles can explain RVH's pathophysiology. The right ventricle adapts to increased afterload from pulmonary hypertension by increasing its contractility and maintaining overall stroke volume and anatomy. In contrast, a failing RV will progressively dilate and functionally decline from the chronically high afterload.
RV cardiac output and stroke volume are functions of the interaction between intrinsic contractility and afterload. The "coupling" of the ventriculoarterial system maintains forward cardiac output, while uncoupling results in RV failure. Chronic afterload elevation in pulmonary hypertension produces compensatory RVH (see Image. Right Ventricular Wall Thickening in Ventricular Hypertrophy).
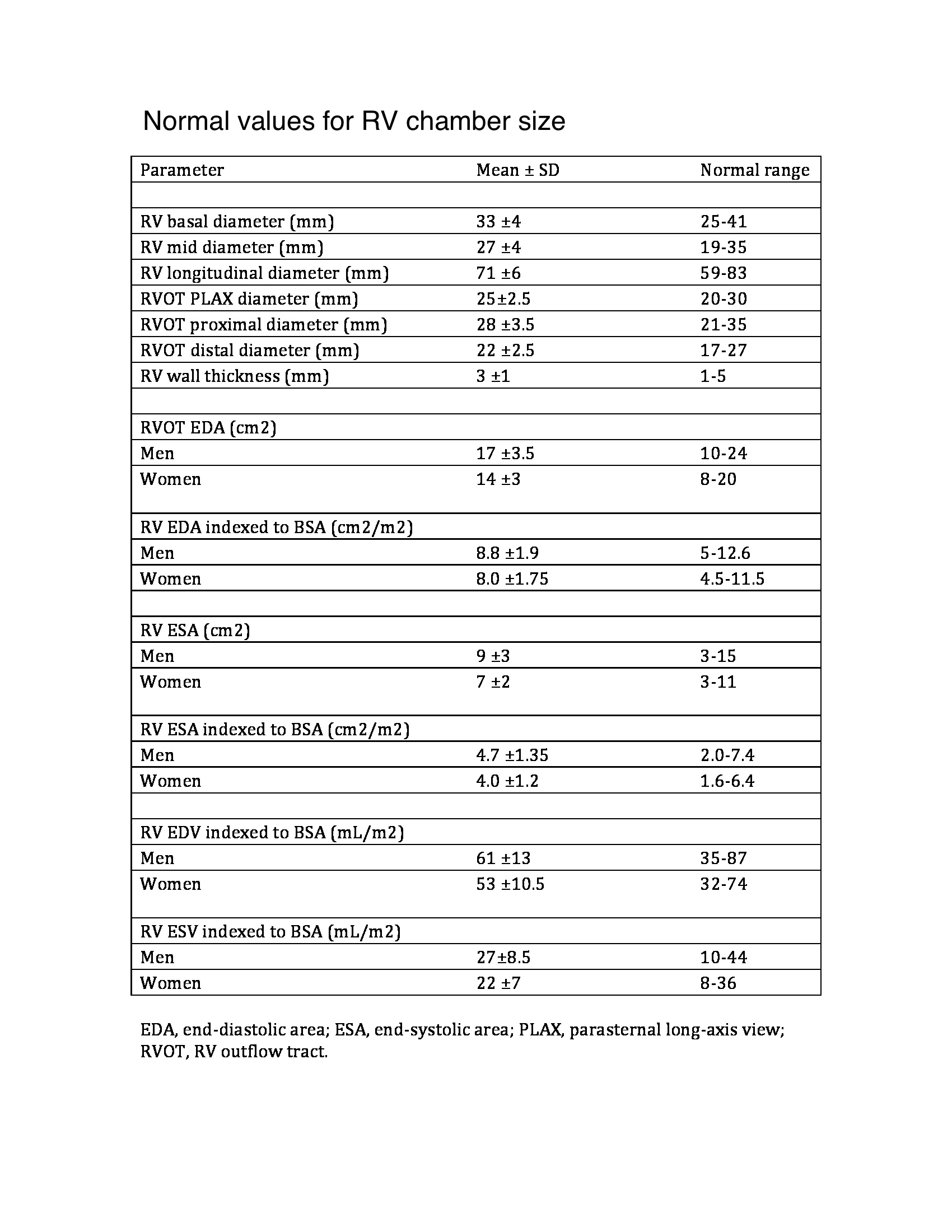
Chronic pulmonary hypertension activates neurohormonal systems, including the sympathetic nervous and renin-angiotensin-aldosterone systems, increasing contractility and hypertrophy (see Image. Normal Values for Right Ventricle Chamber Size). The primary cause of significant pulmonary hypertension is almost always PVR elevation. Increased right ventricular output alone does not usually cause significant pulmonary hypertension because the pulmonary vascular bed vasodilates and recruits vessels in response to increased flow.
Similarly, increased pulmonary venous pressure, represented by the alveolar occlusion pressure, does not usually cause significant pulmonary hypertension. However, a chronic increase in right ventricular output, pulmonary venous pressure, or both can raise PVR.
Numerous medical conditions can alter all variables:
- Increased pulmonary vascular resistance arises from various factors. One is occlusive vasculopathy leading to PAH, characterized by remodeling and altered vascular tone in the small pulmonary arteries and arterioles. Another is hypoxic vasoconstriction from hypoventilation syndromes and parenchymal lung disease. Factors that reduce the pulmonary vascular bed's cross-sectional area, like pulmonary emboli and interstitial lung disease, can also elevate PVR.
- Increased flow through the pulmonary vasculature may be due to congenital heart defects with left-to-right shunting (eg, atrial and ventricular septal defects and patent ductus arteriosus), anemia, and liver cirrhosis.
- Increased pulmonary venous pressure may be due to left ventricular systolic or diastolic dysfunction, mitral valve disease, constrictive or restrictive cardiomyopathy, or pulmonary venous obstruction (eg, pulmonary venoocclusive disease).
Pulmonary hypertension is a proliferative vasculopathy of the small pulmonary muscular arterioles (diameter < 50 μ) characterized by vasoconstriction, hyperplasia, hypertrophy, fibrosis, and thrombosis involving all 3 vascular wall layers (intima, media, and adventitia). Patients with idiopathic PAH and heritable variants may have a genetic predisposition to pulmonary hypertension (eg, bone morphogenetic protein receptor 2 mutations) with additional contributing mechanisms, including vasoactive mediators, potassium channel dysfunction, and abnormal response to estrogen. The pathogenesis of PAH from other etiologies, including drugs and toxins, connective tissue disease, congenital heart disease, HIV, portopulmonary hypertension, schistosomiasis, pulmonary venoocclusive disease, and persistent pulmonary hypertension of the newborn, is poorly understood.
Blood retrograde flow into the right atrium during systole characterizes tricuspid regurgitation. Since the right atrium is relatively compliant, no significant hemodynamic consequences are evident with mild or moderately severe tricuspid regurgitation. In contrast, severe cases lead to right atrial and venous pressure increase, producing right-sided heart failure signs and symptoms. Right ventricular pressure or volume overload leads to systolic dysfunction and a low cardiac output.
History and Physical
History
RVH presents with signs and symptoms of right-sided heart failure. However, the manifestations develop gradually, usually after a longstanding pulmonary cardiac pathology. Patients often consult for acutely worsening symptoms. Chronicity is one attribute that can distinguish RVH from right-sided heart failure arising from acute left ventricular disease.
Exertional chest pain (angina) is often reported and is usually due to subendocardial hypoperfusion from increased myocardial oxygen demand and right ventricular wall stress. This symptom may also arise from the left main coronary artery’s dynamic compression by an enlarged pulmonary artery. This risk is most significant in patients with a pulmonary artery trunk at least 40 mm in diameter.
Peripheral edema may be elicited, arising from right ventricular failure, increased right-sided filling cardiac pressures, and extracellular volume expansion. Patients may also experience exertional syncope due to the inability to augment cardiac output during activity. Reflex bradycardia secondary to right ventricular mechanoreceptor activation also causes exertional syncope. Palpitations are also common.
Anorexia and right upper quadrant (RUQ) abdominal pain may be reported due to passive hepatic congestion. Uncommon symptoms include coughing, hemoptysis, and hoarseness (Ortner syndrome). Hoarseness arises from left recurrent laryngeal nerve compression by a dilated main pulmonary artery.
Patients’ past medical history is usually remarkable for a chronic pulmonary or cardiac condition, such as chronic obstructive pulmonary disease (COPD), tricuspid regurgitation, and chronic left ventricular failure. Genetic pulmonary or cardiac disorders may be elicited in the family history.
Decompensated patients may present unconscious, pulseless, and apneic. These symptoms indicate cardiorespiratory arrest, warranting immediate resuscitation.
Physical Examination
The general examination of patients with RVH often reveals cachexia, cyanosis, and respiratory distress. Oxygen saturation is typically low. Tachycardia may be appreciated with or without arrhythmia. Hypotension in this setting may signify left heart failure and shock.
Jugular vein assessment
Distended and prominent jugular veins are apparent and reflect right atrial pressure elevation. A distinct “c-v” wave in jugular venous pulse measurements is due to systolic regurgitation through the tricuspid valve into the right atrium. RVH also produces a prominent jugular venous pulse “a” wave, often accompanied by a right-sided S4 and either a left parasternal heave or downward subxiphoid thrust. Jugular venous distension may be more prominent with inspiration (Kussmaul sign) due to the increased venous return. The Kussmaul sign may not be evident if jugular venous distension is severe.
Chest palpation
Chest palpation may reveal a dynamic right ventricular heave due to the chamber’s dilatation. A sustained systolic left parasternal lift is most frequently appreciated in the presence of significant RVH. Longstanding, severe PAH, whether precapillary (eg, in idiopathic PAH or pulmonary valve stenosis) or postcapillary (eg, in mitral stenosis or cardiomyopathy), produces RVH and a sustained lower left parasternal lift. Severe PAH may be associated with a palpable presystolic ‘a’ wave preceding the right ventricular heave, suggesting decreased compliance. Epigastric and subxiphoid pulsations are usually abnormal and related to RVH and abdominal aortic aneurysm dilation. However, subxiphoid pulsations may not always indicate RVH in patients with emphysema.
A hyperdynamic but unsustained left parasternal systolic impulse may be palpable with right ventricular volume elevation, as in an atrial septal defect (ASD) or tricuspid regurgitation. The left parasternal impulse is sustained during systole when PAH is also present.
Severe tricuspid regurgitation produces detectable pulsations along the right sternal border due to right atrial reflux. A systolic outward impulse palpable along the left parasternal and midprecordial regions may still be present even without RVH in patients with significant mitral regurgitation. The systolic pulsation results from left atrial expansion due to mitral regurgitation, pressing the anterior structures toward the anterior chest wall. A right parasternal systolic outward movement from a large, ventricularized right atrium may be appreciated in some patients with Ebstein anomaly.
Cardiac auscultation
An S3 is associated with a highly dilated right ventricle and may vary in intensity and inspiration. An S4 may also be heard if significant RVH is appreciated.
A finding indicative of pulmonary hypertension is increased S2 intensity due to its pulmonic component. Patients with preserved right ventricular function may have either a closely split or single S2. Right ventricular failure or a right bundle branch block (RBBB) widens S2 splitting. Other cardiac sounds may be heard if other heart conditions are present.
Tricuspid regurgitation is classically associated with a holosystolic murmur, best heard at the right or left midsternal border or subxiphoid area. The murmur may be appreciated at the apex in the presence of right ventricular enlargement (RVE). The murmur usually has little radiation, and a thrill is not palpable. However, the murmur of tricuspid regurgitation is often soft or absent, even when regurgitation is severe. Diastolic murmurs are usually absent in tricuspid regurgitation. However, a diastolic rumble may be heard in the presence of tricuspid stenosis or substantial blood flow across the tricuspid valve during diastole, as seen in ASD.
Maneuvers that increase venous return, such as leg raising, exercise, or hepatic compression, augment the murmur of tricuspid regurgitation. The murmur may be louder after a premature beat and prolonged diastole. On the other hand, reducing venous return with standing or amyl nitrate administration diminishes the murmur’s intensity.
The murmur’s intensity may change with pulmonary artery pressure fluctuations in patients with pulmonary hypertension. Respiratory variation in the murmur’s intensity and duration (Rivero-Carvallo sign) may be observed in mild to moderate tricuspid regurgitation. Inspiration promotes venous return to the right ventricle, making the murmur louder and longer unless the murmur is holosystolic. Respiratory variation may occasionally be augmented when the patient is standing and venous return is reduced. However, patients with severe tricuspid regurgitation or marked RVE and right heart dysfunction may not exhibit the murmur’s respiratory variation.
Pulmonary auscultation
RVH pulmonary findings are diverse and contingent upon the underlying etiology. RVH secondary to PAH often manifests signs of elevated pulmonary artery pressures, such as prominent pulmonary artery pulsations and an accentuated pulmonary S2 component. Conversely, RVH due to chronic lung diseases like COPD or interstitial lung disease may primarily produce auscultation sounds reflective of the underlying lung condition, such as decreased breath sounds or crackles. Decreased vocal and tactile fremiti may indicate pleural effusion.
Extremity examination
Ascites and peripheral edema of variable severity may be present, and anasarca can occur in severe diseases. Unilateral or bilateral pleural effusions increase in frequency when RVH results from pulmonary hypertension secondary to a left-sided cardiac valvular or myocardial pathology. Some patients with pulmonary hypertension due to severe COPD may develop edema in the absence of right heart failure.
Abdominal assessment
The liver is enlarged and tender and may be pulsatile in severe RVH. A systolic murmur (thrill) may occasionally be heard over the liver. Patients may develop ascites as a manifestation of right heart failure. Jaundice may arise from secondary liver dysfunction.
Evaluation
RVH evaluation focuses on chamber quantification and assessment of the underlying cause and associated cardiac abnormalities.[11][12][13]
Chest Radiography
Chest radiographs of patients with severe RVH reveal cardiomegaly due to RVE. A prominent cardiac silhouette is observed on the right on pulmonary artery view. The enlarged right ventricle fills the retrosternal space on lateral view. Additional findings may include right atrial enlargement (RAE), azygos vein prominence, an upwardly displaced diaphragm, or pleural effusion. RVH from pulmonary hypertension secondary to a left-sided cardiac abnormality is often associated with other radiographic abnormalities, particularly prominent right and left hilar pulmonary artery segments.
Electrocardiography
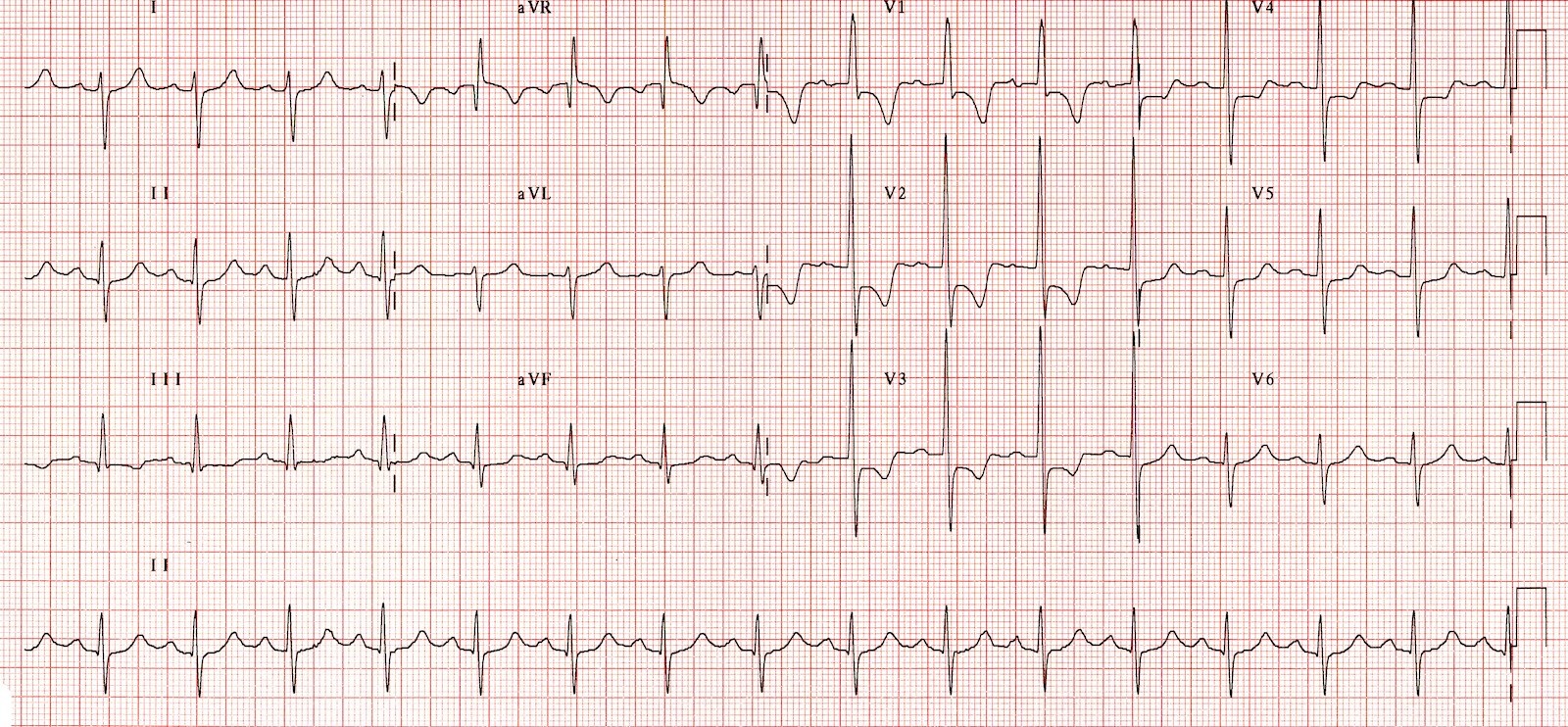
The electrocardiogram (ECG) is indispensable in evaluating RVH. RAD is defined as having the QRS axis shifting between +90° to +180° (normal is between -30° and +90°) and is often a sign of RVH.[21] Associated right atrial overload and ST-segment and T-wave abnormalities (formerly called “right ventricular strain”) may exist in the right precordial leads, reflecting subendocardial ischemia or repolarization abnormalities of the right ventricular myocardium.
Right ventricular forces are predominant in patients with RVH, producing tall R waves in the right precordial leads (V1 and V2) and deep S waves in the left precordial leads (V5 and V6). RVH is often present when the V1 lead has a tall R wave and shallow S wave, making the R-S ratio greater than 1.
The differential diagnosis of an increased R-S ratio in adults includes RBBB, posterior wall myocardial infarction, Wolff-Parkinson-White pattern (especially due to lateral or posterolateral left ventricular preexcitation), hypertrophic cardiomyopathy (particularly due to septal hypertrophy), early precordial transition (counterclockwise rotation), and normal or positional variant. An R-S ratio greater than 1 from a right-sided precordial lead (V3R or V4R) is a more reliable RVH indicator.
Thus, ECG clues to the diagnosis of RVH include the following (see Image. Right Ventricular Hypertrophy Electrocardiography):
- RAD (greater than 90°)
- R in V1 greater than 6 mm
- R in V1 + S in V5 or V6 greater than 10.5 mm
- R-S ratio in V1 greater than 1
- S-R ratio in V6 greater than 1
- Late intrinsicoid deflection in V1 (greater than 0.035 seconds)
- Incomplete RBBB
- ST-T wave abnormalities ("right ventricular strain") in the inferior leads
- RVH or right ventricular overload (“P pulmonale”)
- S greater than R in leads I, II, III, particularly in children (S1, S2, S3 pattern)
Patients with VSD and ECG evidence of RVH require further evaluation to determine the cause, whether pulmonary hypertension, pulmonary stenosis, or a double-chambered right ventricle).
Clinicians should be aware that variations in the accuracy of RVH criteria and conflicting literature findings may arise from different underlying pathophysiologic mechanisms yielding distinct ECG patterns. Tall right precordial R waves with RAD are typically linked to severe RV pressure overloads caused by pulmonic stenosis or pulmonary hypertension not stemming from COPD (eg, primary pulmonary hypertension or recurrent pulmonary emboli). Pulmonary hypertension due to severe COPD (emphysema) may manifest as slow R wave progression, delayed precordial transition zone, and RAD. RVH arising from a classic volume load state, such as ostium secundum ASD, may present with right ventricular conduction delay and RAD.
Echocardiography
Echocardiography can assess RVH by evaluating chamber size, thickness, function, and pulmonary hypertension and tricuspid regurgitation severity. The chamber has 3 walls: anterior, lateral, and inferior. The anterior wall may be assessed on a parasternal long-axis view. The lateral wall can be seen on an apical 4-chamber view. The inferior wall can be seen on a subcostal 4-chamber view. All 3 walls can be appreciated on a parasternal short-axis view at the midventricular level. The normal right ventricular wall thickness range is 3 to 5 mm. Wall thickness greater than 5 mm suggests RVH. Right ventricular wall thickness varies due to trabeculations. Ventricular thickness bears a positive correlation with pulmonary artery pressures. Real-time 3-dimensional echocardiography is superior to conventional 2-dimensional echo for right ventricular mass quantification.
Diagnostic testing is indicated whenever pulmonary hypertension is suspected. Diagnostic testing confirms pulmonary hypertension, determines its severity, and identifies its cause.
An echocardiogram nonindicative of pulmonary hypertension should prompt diagnostic evaluation guided by clinical considerations. If suspicion is low, symptoms should be reassessed for alternative diagnoses. Right heart catheterization (RHC) is warranted if suspicion for pulmonary hypertension persists despite echocardiographic findings.
On the other hand, no further testing is required when an echocardiogram suggests pulmonary hypertension and evidence of left heart disease is apparent. However, further diagnostic testing is necessary in the absence of evidence of left heart disease or when left heart disease appears insufficient to explain the extent of estimated pulmonary hypertension.
Echocardiography is the main diagnostic modality for evaluating tricuspid regurgitation. The operator should examine the right ventricle using multiple acoustic windows, reporting qualitative and quantitative parameters. This modality helps evaluate tricuspid regurgitation severity, valve morphology, right chamber sizes, right ventricular function, pulmonary artery systolic pressure, and the presence of concomitant left heart disease.
Quantifiable parameters include right ventricular and atrial dimensions and systolic function through 1 or a combination of the following:
- Fractional area change
- Doppler tissue imaging-derived tricuspid lateral annular systolic velocity wave
- Tricuspid annular plane systolic excursion
- Right ventricular index of myocardial performance
Right ventricular systolic pressure, typically calculated using the tricuspid regurgitation jet and estimating right atrial pressure based on inferior vena cava size and collapsibility, should be reported when a complete tricuspid regurgitation Doppler velocity envelope is present. Additional parameters such as right ventricular volumes and ejection fraction using 3-dimensional echocardiography should complement the basic 2-dimensional echocardiographic measurements when feasible. The 2015 American Society of Echocardiography guidelines display the new reference values in Tables 2A and 2B.[22]
Cardiovascular Magnetic Resonance
Cardiovascular magnetic resonance (CMR) imaging is the gold standard for evaluating RVH and is especially useful if the echocardiographic evaluation is suboptimal or inconclusive for assessing RVH severity and right ventricular size and function. CMR also enables quantitative assessment of tricuspid regurgitant volume, regurgitant fraction (the ratio of tricuspid regurgitant volume to stroke volume), right ventricular volumes, ejection fraction, and associated left ventricular and mitral disease.
Cardiac Catheterization and Angiography
Cardiac catheterization and contrast right ventriculography do not help diagnose or evaluate RVH in most patients. However, RHC measurement of pulmonary pressures and vascular resistance is appropriate in patients with RVH when clinical and noninvasive data regarding pulmonary pressures are discordant. Left heart catheterization may help assess potential causes of functional tricuspid regurgitation, such as a left-sided valve or myocardial disease with an elevated left atrial pressure. On the other hand, a diagnosis of pulmonary hypertension requires RHC. Pulmonary hypertension is confirmed when the mean pulmonary artery pressure is 25 mm Hg or greater at rest. Clinical studies and additional information provided by RHC are necessary to classify the patient into an appropriate WHO pulmonary hypertension category.
Right Heart Catheterization
RHC confirms the diagnosis of pulmonary hypertension and accurately determines the severity of the hemodynamic derangements. RHC is also helpful in distinguishing pulmonary hypertension due to left heart disease, including cases of systolic dysfunction, diastolic dysfunction, or valvular heart disease.
Other Tests
Other diagnostic tests may be included in the workup if ECG and advanced imaging are not enough to determine RVH's underlying etiology or assess its severity. Thus, the following may be considered, depending on clinical presentation:
Pulmonary function tests: Pulmonary function tests (PFTs) may be performed to identify and characterize underlying lung disease contributing to pulmonary hypertension. PFTs may reveal characteristic obstructive or restrictive lung disease patterns, depending on the underlying pathology. PFTs may also show evidence of impaired gas exchange in patients with pulmonary hypertension.
Overnight oximetry: Overnight oximetry can identify nocturnal oxyhemoglobin desaturation. This condition is common in patients with pulmonary hypertension and may prompt supplemental oxygen therapy during sleep.
Polysomnography: Polysomnography is the gold-standard diagnostic test for sleep-related breathing disorders, such as obstructive sleep apnea. The study should be performed when the clinical suspicion for obstructive sleep apnea is high, or the overnight oximetry results are discordant with clinical expectations.
Exercise testing: Exercise testing is usually performed using the 6-minute walk test, stress echocardiography, or cardiopulmonary exercise testing. The latter can be performed with gas exchange measurements, echocardiography, or RHC.
Ventilation-perfusion scanning: Ventilation-perfusion (V/Q) scanning is the preferred imaging study to evaluate patients for CTEPH. A normal V/Q scan accurately excludes chronic thromboembolic disease with a sensitivity of 96% to 97% and a specificity of 90% to 95%. A V/Q scan suggestive of chronic thromboembolic disease should be confirmed by pulmonary angiography, which can also determine the condition’s extent. V/Q scans are an important part of the diagnostic evaluation because pulmonary hypertension due to chronic thromboembolic disease is potentially reversible with surgery.[14]
Treatment / Management
Pulmonary Hypertension Treatment
Pulmonary hypertension treatment reduces right ventricular afterload by decreasing PVR. Combination treatment is usually preferred over monotherapy. The standard regimen includes endothelin receptor antagonists like bosentan, phosphodiesterase inhibitors such as sildenafil, and prostacyclin analogs like iloprost. The AMBITION trial demonstrated that treatment with combined ambrisentan and tadalafil resulted in a greater reduction in clinical heart failure rates and improved exercise capacity.[14]
Loop diuretics are recommended for volume overload, manifesting as peripheral edema and ascites, in patients with severe tricuspid regurgitation and right-sided heart failure. Aldosterone antagonists may provide additional benefits, particularly in those with hepatic congestion and secondary hyperaldosteronism.
Tricuspid regurgitation is often associated with significant left-sided heart disease in most adults. Treatment should be directed at the primary underlying condition. Heart failure due to left ventricular systolic dysfunction must be treated with β-blockers and agents that inhibit the renin-angiotensin-aldosterone system.
The underlying cause of pulmonary hypertension must be addressed. Oxygen and anticoagulant requirements should be assessed. Patients with persistent pulmonary hypertension, categorized as WHO functional class II, III, or IV despite treatment of the underlying cause, should be referred to a specialized center for evaluation for advanced therapy. No single approach for selecting an agent for advanced treatment is recommended. Factors such as WHO functional class, right ventricular function, hemodynamics, vasoreactivity test results, and patient preferences guide the choice of therapy.[15]
Balloon atrial septostomy can be performed in selected patients as a bridge to definitive surgery or as a palliative procedure. Patients with severe pulmonary hypertension may require lung transplantation, particularly if they exhibit persistent New York Heart Association class IV symptoms despite three months of epoprostenol therapy.
Treatment of Associated Tricuspid Regurgitation
Tricuspid valve surgery is suggested for patients with severe symptomatic tricuspid regurgitation despite medical therapy without severe right ventricular systolic dysfunction. Valve repair is preferred over replacement when feasible. However, valvular repair procedures have a significant recurrence risk. Concomitant tricuspid valve repair is advised for patients undergoing left-sided valve surgery with mild, moderate, or greater functional tricuspid regurgitation. Tricuspid surgery is recommended in patients with severe tricuspid regurgitation with or without symptoms or undergoing surgery for left-sided mitral valve disease.[16] The choice between bioprosthetic and mechanical prosthetic tricuspid valves should be individualized based on patient characteristics. A mechanical valve offers durability but requires anticoagulation to reduce the risk of thrombosis.
Differential Diagnosis
RVH must be differentiated from conditions resembling its signs and symptoms or possessing similar ECG features. Clinical entities that may mimic RVH include the following:
- Left-sided heart failure: Left ventricular systolic and diastolic heart failure are well-known causes of peripheral edema, RUQ pain from hepatic congestion, and syncope due to arrhythmias or insufficient cardiac output. Exertional chest pain may also occur. Left-sided heart failure and RVH can be distinguished via echocardiography or cardiac catheterization.
- Coronary artery disease: Myocardial ischemia is the most common cause of exertional chest pain and syncope, arrhythmias, and congestive heart failure symptoms. Coronary artery disease may be identified by stress testing or cardiac catheterization.
- Budd-Chiari syndrome: This condition arises from thrombosis of the hepatic veins and intrahepatic or suprahepatic inferior vena cava. The syndrome causes peripheral edema due to hepatic venous outflow obstruction and RUQ pain due to hepatic congestion. Doppler ultrasonography, computed tomography, magnetic resonance imaging, and venography can help detect this condition.
- Liver disease: Acute and chronic liver diseases cause peripheral edema, which can manifest with RUQ pain. Liver function testing and RUQ ultrasound can help detect liver disease. Liver biopsy is the gold-standard test for liver disease confirmation, though seldom necessary.
As mentioned previously, RVH has ECG features similar to other conditions, including posterior wall myocardial infarction, Wolff-Parkinson-White pattern, hypertrophic cardiomyopathy, early precordial, and transition. The same ECG patterns may also appear in healthy individuals as normal or positional variants.
Staging
Table 1A. WHO Classification of Pulmonary Hypertension
The WHO classification of pulmonary hypertension categorizes the condition into 5 groups based on underlying etiology and pathophysiology. Each group encompasses distinct clinical conditions with specific diagnostic criteria and treatment approaches. This classification system helps clinicians diagnose pulmonary hypertension and tailor the treatment based on the underlying cause and clinical presentation. The classification is as follows:
Group 1. Pulmonary hypertension from pulmonary arterial hypertension
- Idiopathic PAH
- Heritable PAH
- Genetic conditions presenting with PAH, including BMPR2, ALK-1, ENG, SMAD9, CAV1, KCNK3 mutations
- Drug and toxin-induced
- Associated with:
- Connective tissue disease
- Schistosomiasis
- Congenital heart diseases
- Portal hypertension
- HIV infection
- Pulmonary venoocclusive disease, pulmonary capillary hemangiomatosis, or both
- Persistent pulmonary hypertension of the newborn
- Unknown
Group 2. Pulmonary hypertension due to left heart disease
- Left ventricular systolic dysfunction
- Valvular disease
- Left ventricular diastolic dysfunction
- Congenital or acquired left heart inflow or outflow tract obstruction and congenital cardiomyopathies
Group 3. Pulmonary hypertension due to lung diseases, hypoxia, or both
- Interstitial lung disease
- Chronic obstructive pulmonary disease
- Other pulmonary diseases with a mixed restrictive and obstructive pattern
- Alveolar hypoventilation disorder
- Sleep-disordered breathing
- Developmental lung diseases
- Chronic exposure to high altitudes
Group 4. Chronic thromboembolic pulmonary hypertension (CTEPH)
Group 5. Pulmonary hypertension with unclear multifactorial mechanisms
- Systemic disorders: sarcoidosis, pulmonary histiocytosis, lymphangioleiomyomatosis
- Hematologic disorders: chronic hemolytic anemia, myeloproliferative disorders, splenectomy
- Metabolic disorders: glycogen storage disease, Gaucher disease, thyroid disorders
- Others: tumoral obstruction, fibrosing mediastinitis, chronic renal failure, segmental
Prognosis
The prognosis of pulmonary hypertension is highly variable and depends upon the etiology and symptom severity. Generally, untreated severe pulmonary hypertension and right heart failure have a poor prognosis.[17]
Some evidence relates RVH in patients with pulmonary hypertension to survival. Notably, the right ventricular end-diastolic mass index correlates highly with survival. Each 10% increase in this parameter is associated with a 12% rise in mortality. Hence, right ventricular mass metrics may offer prognostic insights in pulmonary hypertension patients, as RVH occurs earlier than right ventricular dilatation and failure in these patients.
Without therapy, individuals with group 1 pulmonary hypertension generally have worse survival than patients with groups 2 to 5 pulmonary hypertension. With surgical treatment, patients with CTEPH tend to have the best survival. People with chronic lung disease associated with pulmonary hypertension (group 3) have worse survival at one year (80% versus 88%), 3 years (52% versus 72%), and 5 years (38% versus 59%) than patients with PAH. Individuals with group 2 pulmonary hypertension have similar survival rates as people with PAH.
Complications
The structural changes in the heart associated with RVH can disrupt the normal electrical conduction system and produce arrhythmias, including atrial and ventricular fibrillation. These arrhythmias can impair the heart's ability to pump blood effectively, potentially resulting in syncope, palpitations, or even sudden cardiac death if not promptly treated.
Right-sided heart failure is another complication of RVH. The hypertrophied right ventricle struggles to overcome increased pressure or volume overload, compromising its ability to pump blood to the lungs over time. Right heart failure can lead to congestion in the systemic venous circulation, causing peripheral edema, ascites, liver dysfunction, and hepatomegaly. Right-sided heart failure can also involve the left side of the heart if left untreated. Biventricular failure arises, producing severe systemic symptoms and significantly compromising a patient's quality of life and overall prognosis. Prompt RVH identification and treatment are crucial in preventing further cardiac function deterioration and improving outcomes for affected individuals.
RVH treatment can also produce complications, including adverse drug reactions, exacerbation of underlying conditions, complications from surgical interventions (if applicable), and potential risks associated with advanced therapies such as implantable devices or transplantation. Aggressive treatment strategies may also lead to electrolyte imbalances, hemodynamic instability, or injury to other organ systems. Close monitoring and management of these possible sequelae are essential in caring for patients with RVH.
Deterrence and Patient Education
For primary prevention, emphasis is placed on identifying and addressing risk factors that can predispose individuals to RVH. Primary preventive measures thus include managing conditions such as pulmonary hypertension, chronic lung diseases, congenital heart defects, and systemic conditions that can lead to increased right ventricular workload. Lifestyle modifications, such as staying fit, exercising regularly, and avoiding tobacco use, can also significantly prevent RVH. Early detection and management of RVH-associated conditions, such as obstructive sleep apnea, are essential in preventing progression.
Secondary prevention focuses on managing RVH in individuals who have already developed the condition. Secondary prevention thus involves regular cardiac function monitoring through diagnostic tests, such as echocardiography and ECG, and assessment of symptoms and risk factors for disease progression. Treatment strategies may include medications to reduce pulmonary hypertension, optimize heart function, and control symptoms. In some cases, surgical interventions, such as pulmonary valve replacement or correcting congenital heart defects, may be necessary to alleviate pressure or volume overload on the right ventricle. Lifestyle modifications and patient education are also important to empower individuals to manage their condition and actively minimize the risk of complications.
Pearls and Other Issues
RVH often signifies underlying cardiovascular pathology, such as pulmonary hypertension, left-sided heart disease, or chronic lung disease. Evaluation must include a comprehensive assessment of the patient's clinical history and physical examination findings. ECG and echocardiography remain cornerstone diagnostic tools. Additional imaging modalities such as RCH and CMR can ascertain the etiology and severity of RVH.
Treatment strategies aim to address the underlying cause, manage symptoms, and prevent RVH progression to right heart failure, with considerations for advanced therapies in refractory cases. Regular follow-up and multidisciplinary collaboration are crucial for optimizing patient outcomes and quality of life. Close monitoring of right ventricular function, hemodynamics, and therapeutic response is imperative for adjusting treatment regimens and mitigating potential complications.
Enhancing Healthcare Team Outcomes
Right heart failure and RVH are best managed by an interprofessional team comprised of a cardiologist, pulmonologist, intensivist, cardiac surgeon, and primary care physician. Cardiologists play a central role in diagnosing and treating RVH, utilizing ECG, echocardiography, and other imaging modalities to assess disease severity and guide treatment decisions. Pulmonologists contribute their expertise in managing underlying lung diseases that may contribute to RVH. Intensivists provide specialized critical care interventions and coordinate multidisciplinary care in the intensive care setting. Cardiothoracic surgeons may be involved in advanced cases requiring surgical interventions, such as pulmonary thromboendarterectomy or heart valve repair. In the outpatient setting, these patients are managed by primary care clinicians.
Nurses provide comprehensive patient care, offering education on medication adherence, lifestyle modifications, and symptom management while monitoring for potential complications. Pharmacists ensure appropriate medication selection, dosing, and monitoring to optimize therapeutic outcomes while minimizing adverse effects. By leveraging the expertise of an interprofessional team, healthcare providers can deliver comprehensive, patient-centered care to individuals with RVH, addressing multifaceted aspects of the disease and promoting optimal outcomes.[18][19][20]