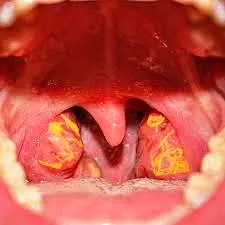
[2]
Ducasa GM,Mitrofanova A,Mallela SK,Liu X,Molina J,Sloan A,Pedigo CE,Ge M,Santos JV,Hernandez Y,Kim JJ,Maugeais C,Mendez AJ,Nair V,Kretzler M,Burke GW,Nelson RG,Ishimoto Y,Inagi R,Banerjee S,Liu S,Szeto HH,Merscher S,Fontanesi F,Fornoni A, ATP-binding cassette A1 deficiency causes cardiolipin-driven mitochondrial dysfunction in podocytes. The Journal of clinical investigation. 2019 Jul 22;
[PubMed PMID: 31329164]
[3]
Schaefer EJ,Kay LL,Zech LA,Brewer HB Jr, Tangier disease. High density lipoprotein deficiency due to defective metabolism of an abnormal apolipoprotein A-i (ApoA-ITangier). The Journal of clinical investigation. 1982 Nov;
[PubMed PMID: 7130397]
[4]
Puntoni M,Sbrana F,Bigazzi F,Sampietro T, Tangier disease: epidemiology, pathophysiology, and management. American journal of cardiovascular drugs : drugs, devices, and other interventions. 2012 Oct 1;
[PubMed PMID: 22913675]
[5]
Serfaty-Lacrosniere C,Civeira F,Lanzberg A,Isaia P,Berg J,Janus ED,Smith MP Jr,Pritchard PH,Frohlich J,Lees RS, Homozygous Tangier disease and cardiovascular disease. Atherosclerosis. 1994 May;
[PubMed PMID: 7945562]
[6]
Mercan M,Yayla V,Altinay S,Seyhan S, Peripheral neuropathy in Tangier disease: A literature review and assessment. Journal of the peripheral nervous system : JPNS. 2018 Jun;
[PubMed PMID: 29582519]
[7]
Kolovou GD,Mikhailidis DP,Anagnostopoulou KK,Daskalopoulou SS,Cokkinos DV, Tangier disease four decades of research: a reflection of the importance of HDL. Current medicinal chemistry. 2006;
[PubMed PMID: 16611066]
[8]
Pervaiz MA,Gau G,Jaffe AS,Saenger AK,Baudhuin L,Ellison J, A Non-classical Presentation of Tangier Disease with Three ABCA1 Mutations. JIMD reports. 2012;
[PubMed PMID: 23430904]
[9]
Pisciotta L,Vitali C,Favari E,Fossa P,Adorni MP,Leone D,Artom N,Fresa R,Calabresi L,Calandra S,Bertolini S, A complex phenotype in a child with familial HDL deficiency due to a novel frameshift mutation in APOA1 gene (apoA-IGuastalla). Journal of clinical lipidology. 2015 Nov-Dec;
[PubMed PMID: 26687706]
[12]
Althaf MM,Almana H,Abdelfadiel A,Amer SM,Al-Hussain TO, Familial lecithin-cholesterol acyltransferase (LCAT) deficiency; a differential of proteinuria. Journal of nephropathology. 2015 Jan;
[PubMed PMID: 25657982]
[13]
Kuivenhoven JA,van Voorst tot Voorst EJ,Wiebusch H,Marcovina SM,Funke H,Assmann G,Pritchard PH,Kastelein JJ, A unique genetic and biochemical presentation of fish-eye disease. The Journal of clinical investigation. 1995 Dec;
[PubMed PMID: 8675648]
[15]
Liang Z,Li W,Yang S,Liu Z,Sun X,Gao X,Yu G, Tangier disease may cause early onset of atherosclerotic cerebral infarction: A case report. Medicine. 2018 Sep;
[PubMed PMID: 30278532]
Level 3 (low-level) evidence
[16]
von Eckardstein A,Huang Y,Assmann G, Physiological role and clinical relevance of high-density lipoprotein subclasses. Current opinion in lipidology. 1994 Dec;
[PubMed PMID: 7712045]
Level 3 (low-level) evidence
[17]
Rondanelli M,Giacosa A,Morazzoni P,Guido D,Grassi M,Morandi G,Bologna C,Riva A,Allegrini P,Perna S, MediterrAsian Diet Products That Could Raise HDL-Cholesterol: A Systematic Review. BioMed research international. 2016;
[PubMed PMID: 27882320]
Level 1 (high-level) evidence
[18]
Yanai H,Tada N, Which Nutritional Factors Are Good for HDL? Journal of clinical medicine research. 2018 Dec;
[PubMed PMID: 30425767]
[19]
Takami H,Kobayashi T,Nakagawa T,Sakurai M,Awata N,Yamashita S, Coronary artery bypass grafting for a patient with Tangier disease. The Japanese journal of thoracic and cardiovascular surgery : official publication of the Japanese Association for Thoracic Surgery = Nihon Kyobu Geka Gakkai zasshi. 2002 Sep
[PubMed PMID: 12382407]
[20]
Muratsu J,Koseki M,Masuda D,Yasuga Y,Tomoyama S,Ataka K,Yagi Y,Nakagawa A,Hamada H,Fujita S,Hattori H,Ohama T,Nishida M,Hiraoka H,Matsuzawa Y,Yamashita S, Accelerated Atherogenicity in Tangier Disease. Journal of atherosclerosis and thrombosis. 2018 Oct 1
[PubMed PMID: 29563393]