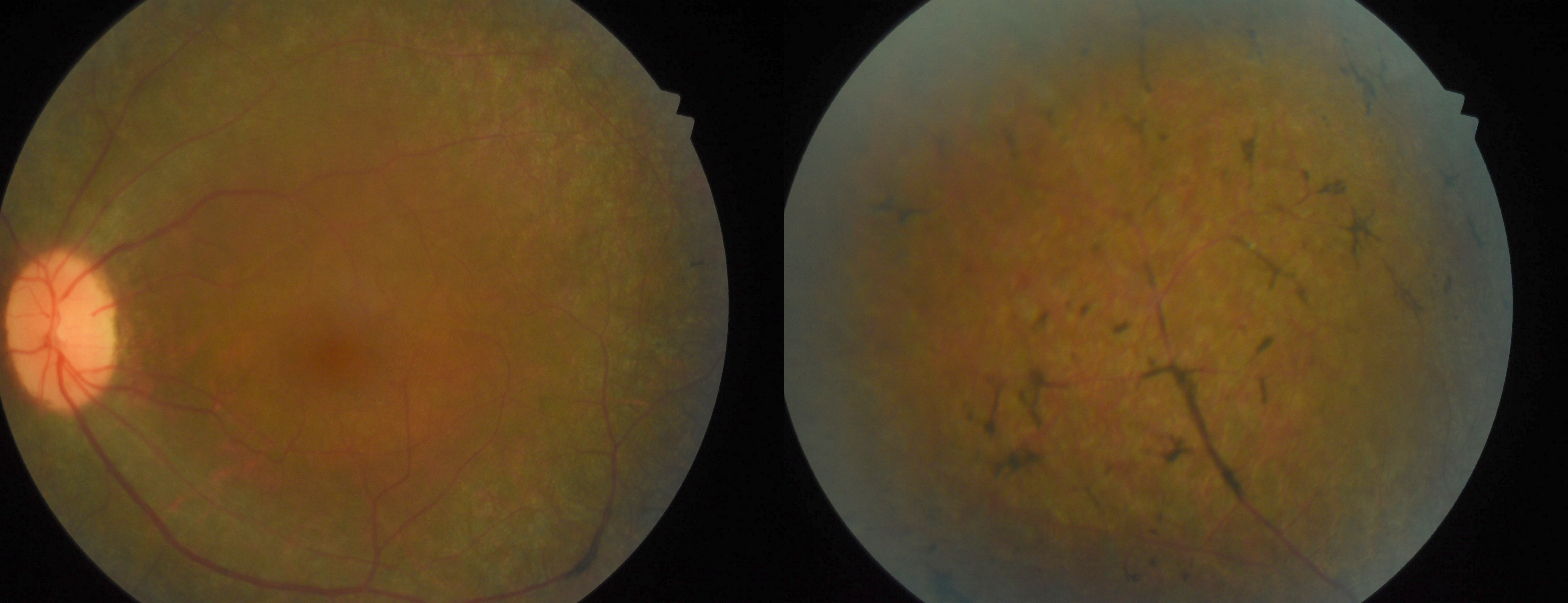
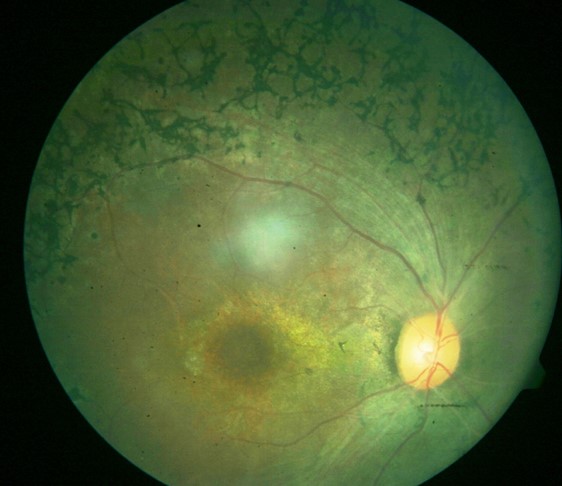
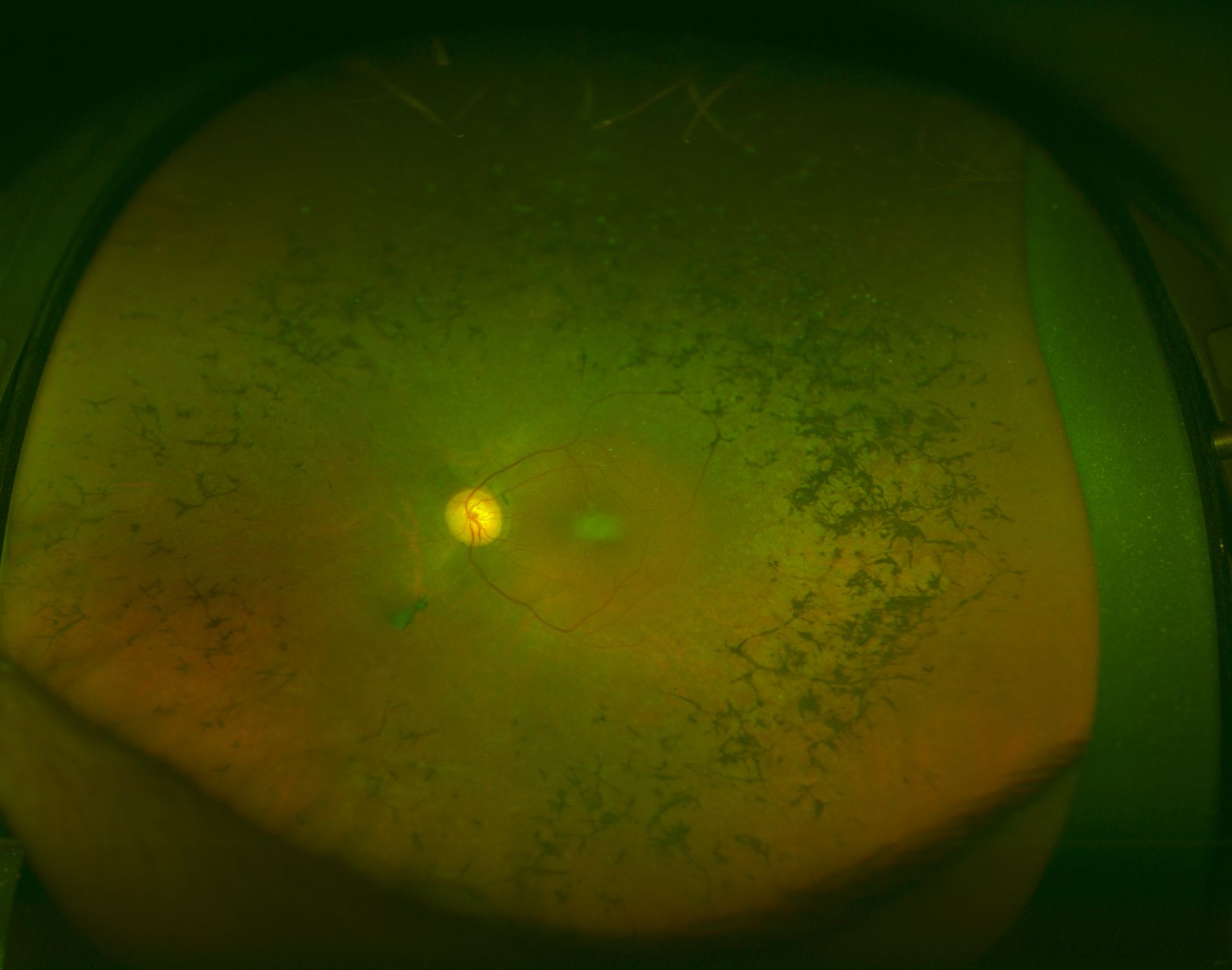
[1]
Verbakel SK, van Huet RAC, Boon CJF, den Hollander AI, Collin RWJ, Klaver CCW, Hoyng CB, Roepman R, Klevering BJ. Non-syndromic retinitis pigmentosa. Progress in retinal and eye research. 2018 Sep:66():157-186. doi: 10.1016/j.preteyeres.2018.03.005. Epub 2018 Mar 27
[PubMed PMID: 29597005]
[2]
Boughman JA, Conneally PM, Nance WE. Population genetic studies of retinitis pigmentosa. American journal of human genetics. 1980 Mar:32(2):223-35
[PubMed PMID: 7386458]
[3]
Weller JM, Michelson G, Juenemann AG. Unilateral retinitis pigmentosa: 30 years follow-up. BMJ case reports. 2014 Feb 10:2014():. doi: 10.1136/bcr-2013-202236. Epub 2014 Feb 10
[PubMed PMID: 24515232]
Level 3 (low-level) evidence
[4]
Bittner AK, Diener-West M, Dagnelie G. Characteristics and possible visual consequences of photopsias as vision measures are reduced in retinitis pigmentosa. Investigative ophthalmology & visual science. 2011 Aug 11:52(9):6370-6. doi: 10.1167/iovs.11-7195. Epub 2011 Aug 11
[PubMed PMID: 21693605]
[5]
O'Hare F, Bentley SA, Wu Z, Guymer RH, Luu CD, Ayton LN. Charles Bonnet Syndrome in Advanced Retinitis Pigmentosa. Ophthalmology. 2015 Sep:122(9):1951-3. doi: 10.1016/j.ophtha.2015.03.006. Epub 2015 Apr 11
[PubMed PMID: 25870080]
[6]
Wolfrum U, Nagel-Wolfrum K. [The Usher Syndrome, a Human Ciliopathy]. Klinische Monatsblatter fur Augenheilkunde. 2018 Mar:235(3):273-280. doi: 10.1055/a-0573-9431. Epub 2018 Mar 13
[PubMed PMID: 29534264]
[7]
Mathur P, Yang J. Usher syndrome: Hearing loss, retinal degeneration and associated abnormalities. Biochimica et biophysica acta. 2015 Mar:1852(3):406-20. doi: 10.1016/j.bbadis.2014.11.020. Epub 2014 Dec 4
[PubMed PMID: 25481835]
[8]
Daiger SP, Sullivan LS, Bowne SJ. Genes and mutations causing retinitis pigmentosa. Clinical genetics. 2013 Aug:84(2):132-41. doi: 10.1111/cge.12203. Epub 2013 Jun 19
[PubMed PMID: 23701314]
[9]
Yang YJ, Peng J, Ying D, Peng QH. A Brief Review on the Pathological Role of Decreased Blood Flow Affected in Retinitis Pigmentosa. Journal of ophthalmology. 2018:2018():3249064. doi: 10.1155/2018/3249064. Epub 2018 Feb 25
[PubMed PMID: 29682340]
[10]
Phelan JK, Bok D. A brief review of retinitis pigmentosa and the identified retinitis pigmentosa genes. Molecular vision. 2000 Jul 8:6():116-24
[PubMed PMID: 10889272]
[11]
Chang S, Vaccarella L, Olatunji S, Cebulla C, Christoforidis J. Diagnostic challenges in retinitis pigmentosa: genotypic multiplicity and phenotypic variability. Current genomics. 2011 Jun:12(4):267-75. doi: 10.2174/138920211795860116. Epub
[PubMed PMID: 22131872]
[12]
Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, Fahim AT, Daiger SP, Weleber RG. Nonsyndromic Retinitis Pigmentosa Overview. GeneReviews(®). 1993:():
[PubMed PMID: 20301590]
Level 3 (low-level) evidence
[13]
Kajiwara K, Berson EL, Dryja TP. Digenic retinitis pigmentosa due to mutations at the unlinked peripherin/RDS and ROM1 loci. Science (New York, N.Y.). 1994 Jun 10:264(5165):1604-8
[PubMed PMID: 8202715]
[14]
Cross N, van Steen C, Zegaoui Y, Satherley A, Angelillo L. Retinitis Pigmentosa: Burden of Disease and Current Unmet Needs. Clinical ophthalmology (Auckland, N.Z.). 2022:16():1993-2010. doi: 10.2147/OPTH.S365486. Epub 2022 Jun 20
[PubMed PMID: 35757022]
[15]
Haim M, Holm NV, Rosenberg T. Prevalence of retinitis pigmentosa and allied disorders in Denmark. I Main results. Acta ophthalmologica. 1992 Apr:70(2):178-86
[PubMed PMID: 1609565]
[16]
Bundey S, Crews SJ. A study of retinitis pigmentosa in the City of Birmingham. I Prevalence. Journal of medical genetics. 1984 Dec:21(6):417-20
[PubMed PMID: 6512829]
[17]
Sen P, Bhargava A, George R, Ve Ramesh S, Hemamalini A, Prema R, Kumaramanickavel G, Vijaya L. Prevalence of retinitis pigmentosa in South Indian population aged above 40 years. Ophthalmic epidemiology. 2008 Jul-Aug:15(4):279-81. doi: 10.1080/09286580802105814. Epub
[PubMed PMID: 18780262]
[18]
Gupta KK, Gurung G, Tulsyan N. Prevalence of Retinitis Pigmentosa in a Tertiary Eye Hospital of Nepal. Nepalese journal of ophthalmology : a biannual peer-reviewed academic journal of the Nepal Ophthalmic Society : NEPJOPH. 2022 Jan:14(27):31-38. doi: 10.3126/nepjoph.v14i1.38977. Epub
[PubMed PMID: 35996901]
[19]
Delmaghani S, El-Amraoui A. The genetic and phenotypic landscapes of Usher syndrome: from disease mechanisms to a new classification. Human genetics. 2022 Apr:141(3-4):709-735. doi: 10.1007/s00439-022-02448-7. Epub 2022 Mar 30
[PubMed PMID: 35353227]
[20]
Tsujikawa M, Wada Y, Sukegawa M, Sawa M, Gomi F, Nishida K, Tano Y. Age at onset curves of retinitis pigmentosa. Archives of ophthalmology (Chicago, Ill. : 1960). 2008 Mar:126(3):337-40. doi: 10.1001/archopht.126.3.337. Epub
[PubMed PMID: 18332312]
[21]
Xu H, Chen M, Forrester JV. Para-inflammation in the aging retina. Progress in retinal and eye research. 2009 Sep:28(5):348-68. doi: 10.1016/j.preteyeres.2009.06.001. Epub 2009 Jun 26
[PubMed PMID: 19560552]
[22]
Arango-Gonzalez B, Trifunović D, Sahaboglu A, Kranz K, Michalakis S, Farinelli P, Koch S, Koch F, Cottet S, Janssen-Bienhold U, Dedek K, Biel M, Zrenner E, Euler T, Ekström P, Ueffing M, Paquet-Durand F. Identification of a common non-apoptotic cell death mechanism in hereditary retinal degeneration. PloS one. 2014:9(11):e112142. doi: 10.1371/journal.pone.0112142. Epub 2014 Nov 13
[PubMed PMID: 25392995]
[23]
Viringipurampeer IA, Gregory-Evans CY, Metcalfe AL, Bashar E, Moritz OL, Gregory-Evans K. Cell Death Pathways in Mutant Rhodopsin Rat Models Identifies Genotype-Specific Targets Controlling Retinal Degeneration. Molecular neurobiology. 2019 Mar:56(3):1637-1652. doi: 10.1007/s12035-018-1192-8. Epub 2018 Jun 18
[PubMed PMID: 29911255]
[24]
Cideciyan AV, Jacobson SG, Aleman TS, Gu D, Pearce-Kelling SE, Sumaroka A, Acland GM, Aguirre GD. In vivo dynamics of retinal injury and repair in the rhodopsin mutant dog model of human retinitis pigmentosa. Proceedings of the National Academy of Sciences of the United States of America. 2005 Apr 5:102(14):5233-8
[PubMed PMID: 15784735]
[25]
Newton F, Megaw R. Mechanisms of Photoreceptor Death in Retinitis Pigmentosa. Genes. 2020 Sep 24:11(10):. doi: 10.3390/genes11101120. Epub 2020 Sep 24
[PubMed PMID: 32987769]
[26]
Wellard J, Lee D, Valter K, Stone J. Photoreceptors in the rat retina are specifically vulnerable to both hypoxia and hyperoxia. Visual neuroscience. 2005 Jul-Aug:22(4):501-7
[PubMed PMID: 16212707]
[27]
B Domènech E, Marfany G. The Relevance of Oxidative Stress in the Pathogenesis and Therapy of Retinal Dystrophies. Antioxidants (Basel, Switzerland). 2020 Apr 23:9(4):. doi: 10.3390/antiox9040347. Epub 2020 Apr 23
[PubMed PMID: 32340220]
[28]
Zhang L, Justus S, Xu Y, Pluchenik T, Hsu CW, Yang J, Duong JK, Lin CS, Jia Y, Bassuk AG, Mahajan VB, Tsang SH. Reprogramming towards anabolism impedes degeneration in a preclinical model of retinitis pigmentosa. Human molecular genetics. 2016 Oct 1:25(19):4244-4255. doi: 10.1093/hmg/ddw256. Epub 2016 Aug 11
[PubMed PMID: 27516389]
[29]
Gallenga CE, Lonardi M, Pacetti S, Violanti SS, Tassinari P, Di Virgilio F, Tognon M, Perri P. Molecular Mechanisms Related to Oxidative Stress in Retinitis Pigmentosa. Antioxidants (Basel, Switzerland). 2021 May 26:10(6):. doi: 10.3390/antiox10060848. Epub 2021 May 26
[PubMed PMID: 34073310]
[30]
Groenendyk J, Agellon LB, Michalak M. Calcium signaling and endoplasmic reticulum stress. International review of cell and molecular biology. 2021:363():1-20. doi: 10.1016/bs.ircmb.2021.03.003. Epub 2021 May 19
[PubMed PMID: 34392927]
[31]
Badano JL, Mitsuma N, Beales PL, Katsanis N. The ciliopathies: an emerging class of human genetic disorders. Annual review of genomics and human genetics. 2006:7():125-48
[PubMed PMID: 16722803]
[32]
Andresen H M, Regueira H T, Leighton F. [Oxidative stress in critically ill patients]. Revista medica de Chile. 2006 May:134(5):649-56
[PubMed PMID: 16802059]
[33]
Milam AH, Li ZY, Fariss RN. Histopathology of the human retina in retinitis pigmentosa. Progress in retinal and eye research. 1998 Apr:17(2):175-205
[PubMed PMID: 9695792]
[34]
Zhao TT, Tian CY, Yin ZQ. Activation of Müller cells occurs during retinal degeneration in RCS rats. Advances in experimental medicine and biology. 2010:664():575-83. doi: 10.1007/978-1-4419-1399-9_66. Epub
[PubMed PMID: 20238061]
Level 3 (low-level) evidence
[35]
Tripathy K, Sharma YR, Chawla R, Basu K, Vohra R, Venkatesh P. Triads in Ophthalmology: A Comprehensive Review. Seminars in ophthalmology. 2017:32(2):237-250. doi: 10.3109/08820538.2015.1045150. Epub 2015 Jul 6
[PubMed PMID: 26148300]
[36]
Shintani K, Shechtman DL, Gurwood AS. Review and update: current treatment trends for patients with retinitis pigmentosa. Optometry (St. Louis, Mo.). 2009 Jul:80(7):384-401. doi: 10.1016/j.optm.2008.01.026. Epub
[PubMed PMID: 19545852]
[37]
Coussa RG, Basali D, Maeda A, DeBenedictis M, Traboulsi EI. Sector retinitis pigmentosa: Report of ten cases and a review of the literature. Molecular vision. 2019:25():869-889
[PubMed PMID: 31908405]
Level 3 (low-level) evidence
[38]
Popović P, Jarc-Vidmar M, Hawlina M. Abnormal fundus autofluorescence in relation to retinal function in patients with retinitis pigmentosa. Graefe's archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie. 2005 Oct:243(10):1018-27
[PubMed PMID: 15906064]
[39]
Robson AG, Egan C, Holder GE, Bird AC, Fitzke FW. Comparing rod and cone function with fundus autofluorescence images in retinitis pigmentosa. Advances in experimental medicine and biology. 2003:533():41-7
[PubMed PMID: 15180246]
Level 3 (low-level) evidence
[40]
Pichi F, Abboud EB, Ghazi NG, Khan AO. Fundus autofluorescence imaging in hereditary retinal diseases. Acta ophthalmologica. 2018 Aug:96(5):e549-e561. doi: 10.1111/aos.13602. Epub 2017 Nov 2
[PubMed PMID: 29098804]
[41]
Robson AG, Lenassi E, Saihan Z, Luong VA, Fitzke FW, Holder GE, Webster AR. Comparison of fundus autofluorescence with photopic and scotopic fine matrix mapping in patients with retinitis pigmentosa: 4- to 8-year follow-up. Investigative ophthalmology & visual science. 2012 Sep 14:53(10):6187-95. doi: 10.1167/iovs.12-10195. Epub 2012 Sep 14
[PubMed PMID: 22899761]
[42]
Lee J, Asano S, Inoue T, Fujino Y, Matsuura M, Kitamoto K, Hashimoto Y, Ogawa A, Yanagisawa M, Azuma K, Murata H, Obata R, Asaoka R. Investigating the Usefulness of Fundus Autofluorescence in Retinitis Pigmentosa. Ophthalmology. Retina. 2018 Oct:2(10):1062-1070. doi: 10.1016/j.oret.2018.03.007. Epub 2018 May 18
[PubMed PMID: 31047495]
[43]
Yung M, Klufas MA, Sarraf D. Clinical applications of fundus autofluorescence in retinal disease. International journal of retina and vitreous. 2016:2():12. doi: 10.1186/s40942-016-0035-x. Epub 2016 Apr 8
[PubMed PMID: 27847630]
[44]
Trichonas G, Traboulsi EI, Ehlers JP. Ultra-widefield fundus autofluorescence patterns in retinitis pigmentosa and other retinal dystrophies. Ophthalmic genetics. 2017 Jan-Feb:38(1):98-100. doi: 10.3109/13816810.2015.1137328. Epub 2016 Apr 6
[PubMed PMID: 27049178]
[45]
Marmor MF. The electroretinogram in retinitis pigmentosa. Archives of ophthalmology (Chicago, Ill. : 1960). 1979 Jul:97(7):1300-4
[PubMed PMID: 454267]
[46]
Grigoropoulos VG, Emfietzoglou J, Nikolaidis P, Chatzistefanou K, Vergados J, Theodossiadis GP, Theodossiadis PG. Optical coherence tomography findings in patients with retinitis pigmentosa and low visual acuity. Ophthalmic surgery, lasers & imaging : the official journal of the International Society for Imaging in the Eye. 2010 Jan-Feb:41(1):35-9. doi: 10.3928/15428877-20091230-07. Epub
[PubMed PMID: 20128568]
[47]
Triolo G, Pierro L, Parodi MB, De Benedetto U, Gagliardi M, Manitto MP, Bandello F. Spectral domain optical coherence tomography findings in patients with retinitis pigmentosa. Ophthalmic research. 2013:50(3):160-4. doi: 10.1159/000351681. Epub 2013 Aug 28
[PubMed PMID: 23989166]
[48]
Takagi S, Hirami Y, Takahashi M, Fujihara M, Mandai M, Miyakoshi C, Tomita G, Kurimoto Y. Optical coherence tomography angiography in patients with retinitis pigmentosa who have normal visual acuity. Acta ophthalmologica. 2018 Aug:96(5):e636-e642. doi: 10.1111/aos.13680. Epub 2018 Mar 1
[PubMed PMID: 29498230]
[49]
Koyanagi Y, Murakami Y, Funatsu J, Akiyama M, Nakatake S, Fujiwara K, Tachibana T, Nakao S, Hisatomi T, Yoshida S, Ishibashi T, Sonoda KH, Ikeda Y. Optical coherence tomography angiography of the macular microvasculature changes in retinitis pigmentosa. Acta ophthalmologica. 2018 Feb:96(1):e59-e67. doi: 10.1111/aos.13475. Epub 2017 May 31
[PubMed PMID: 28561452]
[50]
Chatzinoff A, Nelson E, Stahl N, Clahane A. Eleven-CIS vitamin A in the treatment of retinitis pigmentosa. A negative study. Archives of ophthalmology (Chicago, Ill. : 1960). 1968 Oct:80(4):417-9
[PubMed PMID: 4877320]
[51]
Massoud WH, Bird AC, Perkins ES. Plasma vitamin A and beta-carotene in retinitis pigmentosa. The British journal of ophthalmology. 1975 Apr:59(4):200-4
[PubMed PMID: 1138843]
[52]
Gouras P, Carr RE, Gunkel RD. Retinitis pigmentosa in abetalipoproteinemia: Effects of vitamin A. Investigative ophthalmology. 1971 Oct:10(10):784-93
[PubMed PMID: 5124019]
[53]
Berson EL, Rosner B, Sandberg MA, Hayes KC, Nicholson BW, Weigel-DiFrano C, Willett W. Vitamin A supplementation for retinitis pigmentosa. Archives of ophthalmology (Chicago, Ill. : 1960). 1993 Nov:111(11):1456-9
[PubMed PMID: 8240091]
[54]
Rayapudi S, Schwartz SG, Wang X, Chavis P. Vitamin A and fish oils for retinitis pigmentosa. The Cochrane database of systematic reviews. 2013 Dec 19:2013(12):CD008428. doi: 10.1002/14651858.CD008428.pub2. Epub 2013 Dec 19
[PubMed PMID: 24357340]
Level 1 (high-level) evidence
[55]
Schwartz SG, Wang X, Chavis P, Kuriyan AE, Abariga SA. Vitamin A and fish oils for preventing the progression of retinitis pigmentosa. The Cochrane database of systematic reviews. 2020 Jun 18:6(6):CD008428. doi: 10.1002/14651858.CD008428.pub3. Epub 2020 Jun 18
[PubMed PMID: 32573764]
Level 1 (high-level) evidence
[56]
Piri N, Grodsky JD, Kaplan HJ. Gene therapy for retinitis pigmentosa. Taiwan journal of ophthalmology. 2021 Oct-Dec:11(4):348-351. doi: 10.4103/tjo.tjo_47_21. Epub 2021 Nov 19
[PubMed PMID: 35070662]
[57]
Xi Z, Vats A, Sahel JA, Chen Y, Byrne LC. Gene augmentation prevents retinal degeneration in a CRISPR/Cas9-based mouse model of PRPF31 retinitis pigmentosa. Nature communications. 2022 Dec 13:13(1):7695. doi: 10.1038/s41467-022-35361-8. Epub 2022 Dec 13
[PubMed PMID: 36509783]
[58]
Arsenijevic Y, Berger A, Udry F, Kostic C. Lentiviral Vectors for Ocular Gene Therapy. Pharmaceutics. 2022 Jul 31:14(8):. doi: 10.3390/pharmaceutics14081605. Epub 2022 Jul 31
[PubMed PMID: 36015231]
[59]
Maguire AM, Simonelli F, Pierce EA, Pugh EN Jr, Mingozzi F, Bennicelli J, Banfi S, Marshall KA, Testa F, Surace EM, Rossi S, Lyubarsky A, Arruda VR, Konkle B, Stone E, Sun J, Jacobs J, Dell'Osso L, Hertle R, Ma JX, Redmond TM, Zhu X, Hauck B, Zelenaia O, Shindler KS, Maguire MG, Wright JF, Volpe NJ, McDonnell JW, Auricchio A, High KA, Bennett J. Safety and efficacy of gene transfer for Leber's congenital amaurosis. The New England journal of medicine. 2008 May 22:358(21):2240-8. doi: 10.1056/NEJMoa0802315. Epub 2008 Apr 27
[PubMed PMID: 18441370]
[60]
Dias MF, Joo K, Kemp JA, Fialho SL, da Silva Cunha A Jr, Woo SJ, Kwon YJ. Molecular genetics and emerging therapies for retinitis pigmentosa: Basic research and clinical perspectives. Progress in retinal and eye research. 2018 Mar:63():107-131. doi: 10.1016/j.preteyeres.2017.10.004. Epub 2017 Oct 31
[PubMed PMID: 29097191]
Level 3 (low-level) evidence
[61]
Cross N, van Steen C, Zegaoui Y, Satherley A, Angelillo L. Current and Future Treatment of Retinitis Pigmentosa. Clinical ophthalmology (Auckland, N.Z.). 2022:16():2909-2921. doi: 10.2147/OPTH.S370032. Epub 2022 Aug 31
[PubMed PMID: 36071725]
[62]
Sharma A, Jaganathan BG. Stem Cell Therapy for Retinal Degeneration: The Evidence to Date. Biologics : targets & therapy. 2021:15():299-306. doi: 10.2147/BTT.S290331. Epub 2021 Jul 27
[PubMed PMID: 34349498]
[63]
He Y, Zhang Y, Liu X, Ghazaryan E, Li Y, Xie J, Su G. Recent advances of stem cell therapy for retinitis pigmentosa. International journal of molecular sciences. 2014 Aug 20:15(8):14456-74. doi: 10.3390/ijms150814456. Epub 2014 Aug 20
[PubMed PMID: 25141102]
Level 3 (low-level) evidence
[64]
Limoli PG, Vingolo EM, Limoli C, Nebbioso M. Stem Cell Surgery and Growth Factors in Retinitis Pigmentosa Patients: Pilot Study after Literature Review. Biomedicines. 2019 Nov 30:7(4):. doi: 10.3390/biomedicines7040094. Epub 2019 Nov 30
[PubMed PMID: 31801246]
Level 3 (low-level) evidence
[65]
Smith LE. Bone marrow-derived stem cells preserve cone vision in retinitis pigmentosa. The Journal of clinical investigation. 2004 Sep:114(6):755-7
[PubMed PMID: 15372096]
[66]
Surendran H, Nandakumar S, Reddy K VB, Stoddard J, Mohan K V, Upadhyay PK, McGill TJ, Pal R. Transplantation of retinal pigment epithelium and photoreceptors generated concomitantly via small molecule-mediated differentiation rescues visual function in rodent models of retinal degeneration. Stem cell research & therapy. 2021 Jan 19:12(1):70. doi: 10.1186/s13287-021-02134-x. Epub 2021 Jan 19
[PubMed PMID: 33468244]
[67]
Hill D, Compagnoni C, Cordeiro MF. Investigational neuroprotective compounds in clinical trials for retinal disease. Expert opinion on investigational drugs. 2021 May:30(5):571-577. doi: 10.1080/13543784.2021.1896701. Epub 2021 Apr 1
[PubMed PMID: 33641585]
Level 3 (low-level) evidence
[68]
Birch DG, Bennett LD, Duncan JL, Weleber RG, Pennesi ME. Long-term Follow-up of Patients With Retinitis Pigmentosa Receiving Intraocular Ciliary Neurotrophic Factor Implants. American journal of ophthalmology. 2016 Oct:170():10-14. doi: 10.1016/j.ajo.2016.07.013. Epub 2016 Jul 25
[PubMed PMID: 27457255]
[69]
Chow AY, Bittner AK, Pardue MT. The artificial silicon retina in retinitis pigmentosa patients (an American Ophthalmological Association thesis). Transactions of the American Ophthalmological Society. 2010 Dec:108():120-54
[PubMed PMID: 21212852]
[70]
Gekeler K, Bartz-Schmidt KU, Sachs H, MacLaren RE, Stingl K, Zrenner E, Gekeler F. Implantation, removal and replacement of subretinal electronic implants for restoration of vision in patients with retinitis pigmentosa. Current opinion in ophthalmology. 2018 May:29(3):239-247. doi: 10.1097/ICU.0000000000000467. Epub
[PubMed PMID: 29528862]
Level 3 (low-level) evidence
[71]
Hallum LE, Dakin SC. Retinal Implantation of Electronic Vision Prostheses to Treat Retinitis Pigmentosa: A Systematic Review. Translational vision science & technology. 2021 Aug 12:10(10):8. doi: 10.1167/tvst.10.10.8. Epub
[PubMed PMID: 34383874]
Level 1 (high-level) evidence
[72]
Ward J, Meijer P. Visual experiences in the blind induced by an auditory sensory substitution device. Consciousness and cognition. 2010 Mar:19(1):492-500. doi: 10.1016/j.concog.2009.10.006. Epub 2009 Dec 1
[PubMed PMID: 19955003]
[73]
Grant CA, Berson EL. Treatable forms of retinitis pigmentosa associated with systemic neurological disorders. International ophthalmology clinics. 2001 Winter:41(1):103-10
[PubMed PMID: 11198137]
[74]
Dessalces E, Bocquet B, Bourien J, Zanlonghi X, Verdet R, Meunier I, Hamel CP. Early-onset foveal involvement in retinitis punctata albescens with mutations in RLBP1. JAMA ophthalmology. 2013 Oct:131(10):1314-23. doi: 10.1001/jamaophthalmol.2013.4476. Epub
[PubMed PMID: 23929416]
[75]
Hipp S, Zobor G, Glöckle N, Mohr J, Kohl S, Zrenner E, Weisschuh N, Zobor D. Phenotype variations of retinal dystrophies caused by mutations in the RLBP1 gene. Acta ophthalmologica. 2015 Jun:93(4):e281-6. doi: 10.1111/aos.12573. Epub 2014 Nov 27
[PubMed PMID: 25429852]
[76]
Kurata K, Hosono K, Hotta Y. Long-Term Clinical Course in a Patient with Complete Congenital Stationary Night Blindness. Case reports in ophthalmology. 2017 Jan-Apr:8(1):237-244. doi: 10.1159/000462961. Epub 2017 Apr 10
[PubMed PMID: 28512427]
Level 3 (low-level) evidence
[77]
Zeitz C, Friedburg C, Preising MN, Lorenz B. [Overview of Congenital Stationary Night Blindness with Predominantly Normal Fundus Appearance]. Klinische Monatsblatter fur Augenheilkunde. 2018 Mar:235(3):281-289. doi: 10.1055/s-0043-123072. Epub 2018 Feb 1
[PubMed PMID: 29390235]
Level 3 (low-level) evidence
[78]
Agarwal R, Tripathy K, Bandyopadhyay G, Basu K. Mizuo-Nakamura phenomenon in an Indian male. Clinical case reports. 2019 Feb:7(2):401-403. doi: 10.1002/ccr3.1990. Epub 2019 Jan 13
[PubMed PMID: 30847219]
Level 3 (low-level) evidence
[80]
Thakur A, Puri L. Unilateral retinitis pigmentosa. Clinical & experimental optometry. 2010 Mar:93(2):102-4. doi: 10.1111/j.1444-0938.2009.00435.x. Epub
[PubMed PMID: 20406260]
[81]
Mukhopadhyay R, Holder GE, Moore AT, Webster AR. Unilateral retinitis pigmentosa occurring in an individual with a germline mutation in the RP1 gene. Archives of ophthalmology (Chicago, Ill. : 1960). 2011 Jul:129(7):954-6. doi: 10.1001/archophthalmol.2011.171. Epub
[PubMed PMID: 21746989]
[82]
Tripathy K, Chawla R, Meena S, Agarwal P. Unilateral giant peripapillary drusen and retinal drusenoid deposits in a case of X-linked retinoschisis. BMJ case reports. 2016 Feb 23:2016():. doi: 10.1136/bcr-2016-214558. Epub 2016 Feb 23
[PubMed PMID: 26907824]
Level 3 (low-level) evidence
[83]
Carr RE, Siegel IM. Unilateral retinitis pigmentosa. Archives of ophthalmology (Chicago, Ill. : 1960). 1973 Jul:90(1):21-6
[PubMed PMID: 4714794]
[84]
Toms M, Pagarkar W, Moosajee M. Usher syndrome: clinical features, molecular genetics and advancing therapeutics. Therapeutic advances in ophthalmology. 2020 Jan-Dec:12():2515841420952194. doi: 10.1177/2515841420952194. Epub 2020 Sep 17
[PubMed PMID: 32995707]
Level 3 (low-level) evidence
[85]
Tripathy K, Chawla R, Sarkar S. Girl with polydactyly and pigmentary retinopathy. Journal of paediatrics and child health. 2017 Apr:53(4):424. doi: 10.1111/jpc.1_13323. Epub
[PubMed PMID: 28370859]
[87]
Berson EL. Long-term visual prognoses in patients with retinitis pigmentosa: the Ludwig von Sallmann lecture. Experimental eye research. 2007 Jul:85(1):7-14
[PubMed PMID: 17531222]
[88]
Menghini M, Cehajic-Kapetanovic J, MacLaren RE. Monitoring progression of retinitis pigmentosa: current recommendations and recent advances. Expert opinion on orphan drugs. 2020:8(2-3):67-78. doi: 10.1080/21678707.2020.1735352. Epub 2020 Mar 2
[PubMed PMID: 32231889]
Level 3 (low-level) evidence
[89]
Fahim A. Retinitis pigmentosa: recent advances and future directions in diagnosis and management. Current opinion in pediatrics. 2018 Dec:30(6):725-733. doi: 10.1097/MOP.0000000000000690. Epub
[PubMed PMID: 30234647]
Level 3 (low-level) evidence
[90]
Otsuka Y, Oishi A, Miyata M, Oishi M, Hasegawa T, Numa S, Ikeda HO, Tsujikawa A. Wavelength of light and photophobia in inherited retinal dystrophy. Scientific reports. 2020 Sep 9:10(1):14798. doi: 10.1038/s41598-020-71707-2. Epub 2020 Sep 9
[PubMed PMID: 32908200]
[91]
Watanabe K, Aouadj C, Hiratsuka Y, Yamamoto S, Murakami A. Quality of Life and Economic Impacts of Retinitis Pigmentosa on Japanese Patients: A Non-interventional Cross-sectional Study. Advances in therapy. 2023 May:40(5):2375-2393. doi: 10.1007/s12325-023-02446-9. Epub 2023 Mar 22
[PubMed PMID: 36947329]
Level 2 (mid-level) evidence
[92]
Najjar DM, Igbre AO, Tsai FF. Late capsular bag contraction and intraocular lens subluxation in retinitis pigmentosa: a case report. Journal of medical case reports. 2011 Feb 14:5():65. doi: 10.1186/1752-1947-5-65. Epub 2011 Feb 14
[PubMed PMID: 21320335]
Level 3 (low-level) evidence
[93]
Tripathy K, Chawla R, Venkatesh P, Vohra R, Sharma YR, Gogia V, Jain S, Behera A. Ultra-wide Field Fluorescein Angiography in Retinitis Pigmentosa with Intermediate Uveitis. Journal of ophthalmic & vision research. 2016 Apr-Jun:11(2):237-9. doi: 10.4103/2008-322X.183929. Epub
[PubMed PMID: 27413510]
[95]
Tripathy K. Commentary: Posterior subtenon triamcinolone - The unsung hero for managing various ocular disorders. Indian journal of ophthalmology. 2023 Jan:71(1):181-182. doi: 10.4103/ijo.IJO_1963_22. Epub
[PubMed PMID: 36588232]
Level 3 (low-level) evidence
[96]
Tripathy K. Cystoid Macular Edema in Retinitis Pigmentosa with Intermediate Uveitis Responded Well to Oral and Posterior Subtenon Steroid. Seminars in ophthalmology. 2018:33(4):492-493. doi: 10.1080/08820538.2017.1303521. Epub 2017 Mar 29
[PubMed PMID: 28353369]
[97]
Malik A, Sood S, Narang S. Successful treatment of choroidal neovascular membrane in retinitis pigmentosa with intravitreal bevacizumab. International ophthalmology. 2010 Aug:30(4):425-8. doi: 10.1007/s10792-009-9337-4. Epub 2010 Jan 5
[PubMed PMID: 20049508]
[98]
Sieving PA, Fishman GA. Refractive errors of retinitis pigmentosa patients. The British journal of ophthalmology. 1978 Mar:62(3):163-7
[PubMed PMID: 638108]
[99]
Pradhan C, Khadka S, Joshi P. Angle Closure Glaucoma in Retinitis Pigmentosa. Case reports in ophthalmological medicine. 2020:2020():6023586. doi: 10.1155/2020/6023586. Epub 2020 May 29
[PubMed PMID: 32551144]
Level 3 (low-level) evidence
[100]
Hung MC, Chen YY. Patients with Retinitis Pigmentosa May Have a Higher Risk of Developing Open-Angle Glaucoma. Journal of ophthalmology. 2022:2022():9719095. doi: 10.1155/2022/9719095. Epub 2022 Jun 22
[PubMed PMID: 35783342]
[101]
Badeeb O, Trope G, Musarella M. Primary angle closure glaucoma and retinitis pigmentosa. Acta ophthalmologica. 1993 Dec:71(6):727-32
[PubMed PMID: 8154244]
[102]
Peng DW. [Retinitis pigmentosa associated with glaucoma]. [Zhonghua yan ke za zhi] Chinese journal of ophthalmology. 1991 Sep:27(5):262-4
[PubMed PMID: 1815915]
[103]
Chan WO, Brennan N, Webster AR, Michaelides M, Muqit MMK. Retinal detachment in retinitis pigmentosa. BMJ open ophthalmology. 2020:5(1):e000454. doi: 10.1136/bmjophth-2020-000454. Epub 2020 Jul 9
[PubMed PMID: 32671228]
Level 2 (mid-level) evidence
[104]
Moinuddin O, Sathrasala S, Jayasundera KT, Branham KH, Chang EY, Qian CX, Recchia FM, Fahim AT, Besirli CG. Coats-like Exudative Vitreoretinopathy in Retinitis Pigmentosa: Ocular Manifestations and Treatment Outcomes. Ophthalmology. Retina. 2021 Jan:5(1):86-96. doi: 10.1016/j.oret.2020.03.026. Epub 2020 Apr 9
[PubMed PMID: 32507488]
Level 2 (mid-level) evidence
[106]
Nao-i N, Fukiyama J, Sawada A. Retinitis pigmentosa with recurrent vitreous hemorrhage. Acta ophthalmologica Scandinavica. 1996 Oct:74(5):509-12
[PubMed PMID: 8950405]
[107]
Anil K, Garip G. Coping strategies, vision-related quality of life, and emotional health in managing retinitis pigmentosa: a survey study. BMC ophthalmology. 2018 Jan 30:18(1):21. doi: 10.1186/s12886-018-0689-2. Epub 2018 Jan 30
[PubMed PMID: 29378559]
Level 2 (mid-level) evidence
[108]
Oishi A, Noda K, Birtel J, Miyake M, Sato A, Hasegawa T, Miyata M, Numa S, Charbel Issa P, Tsujikawa A. Effect of smoking on macular function and retinal structure in retinitis pigmentosa. Brain communications. 2020:2(2):fcaa117. doi: 10.1093/braincomms/fcaa117. Epub 2020 Jul 23
[PubMed PMID: 33134916]
[109]
Le P, Nguyen M, Vu T, Dao DP, Olson D, Zhang AY. Anxiety and Depression in Patients With Retinitis Pigmentosa. Journal of vitreoretinal diseases. 2021 Mar-Apr:5(2):114-120. doi: 10.1177/2474126420936455. Epub 2020 Aug 18
[PubMed PMID: 37009075]