Continuing Education Activity
Nevus flammeus or port-wine stain is a capillary malformation usually presenting as a unilateral pink or red discoloration on the skin. These birthmarks, which can vary in size and shape, typically develop at birth or shortly afterward and can occur anywhere on the body of a newborn, although they are most commonly found on the face and neck. Over time, they may grow in size, darken, or become thicker. The color intensity can also change with age. Nevus flammeus is a benign condition that often occurs alone and is only of cosmetic concern. However, when it is a part of a combined vascular malformation or a syndrome, the patient will require further evaluation, including radiological imaging, to confirm the diagnosis. Initiating appropriate treatment is crucial to prevent the development/ progress of any disabilities and prompt diagnosis of any anticipated complications/ visceral anomalies. This activity reviews the evaluation and treatment of nevus flammeus and highlights the role of an interprofessional team in the management of affected patients.
Objectives:
Identify the pathophysiology of nevus flammeus.
Assess the typical clinical presentation and syndromic associations of nevus flammeus.
Implement evidence-based treatment strategies to manage nevus flammeus and minimize potential complications.
Collaborate with an interprofessional team to provide comprehensive care for patients with nevus flammeus.
Introduction
Nevus flammeus or port-wine stain (PWS) is a non-neoplastic congenital dermal capillary hamartomatous malformation presenting as a pink or red patch on a newborn's skin. It is a congenital skin condition that can affect any part of the body and persists throughout life.[1] The nevus flammeus is a well-defined, often unilateral, bilateral, or centrally positioned pink to red patch that appears on the face at birth and is made up of distorted capillary-like vessels.[2] It needs to be differentiated from a nevus simplex/salmon patch, which is usually seen along the midline and disappears over time.[3] An acquired port-wine stain, clinically and histopathologically indistinguishable from congenital capillary malformation, has been reported to develop in adolescents or adults, usually following trauma.
Although nevus flammeus is a benign lesion, and often the diagnosis is clinical, occasionally detailed evaluation and radiological imaging are required when associated with other skin, soft tissue, skeletal, and vascular anomalies.
Etiology
The etiology of nevus flammeus is yet to be clearly defined. Most cases are sporadic; however, familial cases have also been reported.[1] The advances made in the field of genetics have been instrumental in determining the genetic basis of some of the syndromic and non-syndromic forms of nevus flammeus. A somatic activating mutation in GNAQ (located on chromosome 9; locus 9q21.2) has been demonstrated in isolated nevus flammeus and Sturge-Weber syndrome (SWS).[1]
Mutations, more commonly in RASA1 and rarely in EPHB4, have been identified in cases of capillary malformation-arteriovenous malformation syndrome.[4] PIK3CA-related overgrowth spectrum (PROS), which includes CLOVES syndrome, Klippel-Trenaunay syndrome, and macrocephaly-capillary malformation, is caused by postzygotic mosaic activating mutations in PIK3CA.[5] Proteus syndrome is also an overgrowth syndrome secondary to somatic activating mosaic mutations in AKT1.[6] GNA11 mutation has been reported to cause diffuse capillary malformation with overgrowth.[7]
Neuronal dysregulation and overexpression of vascular endothelial growth factors (VEGF) and their components have also been proposed as etiological factors for developing nevus flammeus.[8] An acquired nevus flammeus has been reported very rarely following trauma and is referred to as "Fegeler syndrome."
Epidemiology
A port-wine stain (PWS) is one of the most common vascular anomalies. It almost always presents at birth, affecting 0.3% to 0.5% of newborns, often in the head and neck region. No gender predilection has been noted.[1][9]
Pathophysiology
Understanding the ultrastructural changes and molecular profile of endothelial cells of blood vessels in the affected skin and uncovering the genetic basis of vascular malformations have been crucial to better comprehending this congenital skin condition. Congenital weakness of vessel walls, defects in perivascular dermal elements, abnormalities in neuromodulation, and dysmorphogenesis of cephalic neuroectoderm are some theories used to explain the origin of this vascular malformation.[3]
The 2 major hypotheses, with evidence from recent studies in their favor, are defective nerve innervations to dermal blood vessels and genetic mutations leading to dysregulation of angiogenic signaling.[9] Vessels in PWS lack normal nerve innervations, as evident from the significant decrease in S-100 positive nerve fibers, affecting only the vessels in the dermis.[9] Consequently, there is a decrease in the basal tone of the vessel and/or loss of neurotrophic factors, resulting in the pathological changes of PWS.[6] It has been proposed that there is a defective maturation of the sympathetic fibers in the dermal vessels that happens embryologically, which results in the loss of sympathetic control, which in turn leads to vascular ectasia.[10]
Dermal blood vessels in PWS coexpress both arterial marker (EfnB2) and venous marker (EphB1), which lead to aberrations in the normal differentiation of the primary capillary plexus into dermal arterioles and venules. The result is a venule-like vasculature that undergoes progressive dilatation. Abnormal activation of mitogen-activated protein kinases (MAPK) and phosphoinositide 3-kinase (PI3K) signaling pathways, which play a key role in angiogenesis, have also been implicated.[9] Mutations in RASA1, GNAQ, EphB4, and EphB1/ EfnB2 coexpression are some of the inciting factors for this abnormal activation.
The causes for abnormal and dilated blood vessels in PWS can be summarized as follows:[11]
- Somatic GNAQ (R183Q) mutations that cause angiopoietin-2 lead to the formation of enlarged capillary-like vessels
- Reduced number of perivascular nerve elements
- Coexistence of Eph receptor B1 and ephrin B2
- Absent expression of alpha-smooth muscle actin
- Novel mutations like PIK3CA, SMARCA4, EPHA3, MYB, and PDGFR-beta
Histopathology
Dilated, ectatic capillaries and postcapillary venules of variable calibers in the papillary and upper reticular dermis are seen in the histology of a PWS. There is no proliferation of endothelial cells or vessels.[1] With age, progressive vascular dilatation will be seen with or without perivascular fibrosis, which is clinically evident as hypertrophy and nodularity developing during the natural evolution of PWS.
History and Physical
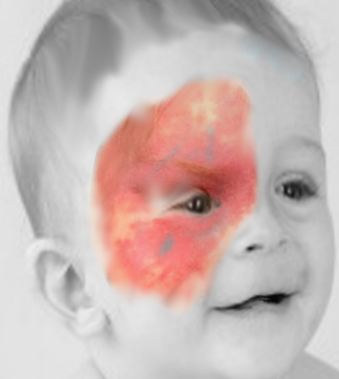
PWS is the most common vascular malformation, typically presenting as pink or red homogenous, variably sized macules and patches with geographic borders at birth that persist throughout life. It is painless, does not bleed spontaneously, and is not warm to touch. In 70% to 90% of cases, it affects the skin over the head and neck; however, it can be seen over the trunk or extremities and can rarely involve the mucosa.[1][9][12] The lesions can be single or multiple, unilateral or bilateral, localized or widespread. Over the face, it is usually segmental, conforming to the trigeminal nerve distribution (see Image. Port Wine Stain). It increases in size proportionate to the physical growth of the child. With age, the lesions darken and acquire a purple hue. Thickening and hypertrophy can be seen most commonly with facial PWS later in life with or without nodularity due to progressive ectasia of dermal vessels. Patients can rarely develop hyperplasia of underlying fat, muscle, and bone, predominantly with facial PWS.[1][9]
PWS over the midline lumbosacral area could be a cutaneous marker of occult spinal dysraphism. Imaging studies are indicated when PWS is associated with other cutaneous markers like a tuft of hair, hemangioma, lipoma, dermoid cyst, and true/pseudo tail.[13]
In 1949, Fegeler described skin lesions that were morphologically and histopathologically similar to PWS developing following trauma. This infrequently reported clinical entity, which shows a better response to pulsed dye laser, is called Fegeler syndrome or acquired PWS. The commonly reported trigger is trauma; other triggers that have been described are the use of drugs like isotretinoin, oral contraceptive pills, simvastatin, metformin, frostbite injury, herpes zoster infection, and obstruction of a peritoneovenous shunt.[14]
PWS often occurs as an isolated vascular malformation; however, in an individual with other congenital disabilities, it could be a part of a genetic syndrome. Some of these syndromes, with a brief description of their hallmark clinical features, are listed below.
Sturge-Weber Syndrome (SWS)
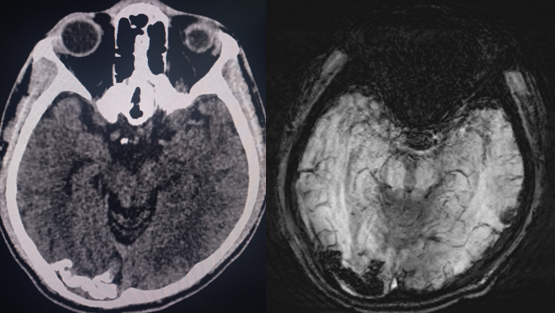
Sturge-Weber syndrome is a neurocutaneous syndrome characterized by PWS, leptomeningeal angiomatosis, and/or eye involvement. The PWS is usually unilateral and along the distribution of the trigeminal nerve. However, it can occasionally be bilateral, extensive, and sometimes involve the trunk and the extremities. Children with SWS often develop seizures by the age of 2 years that can be associated with contralateral neurological deficits and learning difficulties. Imaging of the brain may show cerebral atrophy with gyriform calcifications, described as a "tram-track sign" (see Image. Sturge-Weber Syndrome). In the eye, choroid angioma can lead to complications like glaucoma, buphthalmos, and retinal detachment.[1][6]
A facial nevus flammeus, including any area of the forehead, including the upper eyelid and the midline frontonasal prominence, is the greatest indicator of SWS. The long-held notion that the distribution has a "trigeminal nerve" etiology is challenged by the observation that it appears to follow the patterns of embryological vasculature. Notably, not all patients with nevus flammeus will develop SWS; however, forehead location is the strongest independent predictor.
High-risk factors for associated central nervous system abnormalities in a patient with PWS: [15]
- Extensive bilateral involvement
- Hemifacial involvement of the forehead
- Median PWS
- >50% of contiguous hemi-forehead involvement
Klippel-Trenaunay Syndrome (KTS)
Klippel-Trenaunay syndrome is a sporadic condition characterized by a combined capillary-lymphatic-venous malformation with bone and soft tissue hypertrophy. The lower limbs are the most common site involved. Persistence of the embryonic vein of the lateral thigh (vein of Servelle) and stenosis/hypoplasia/aplasia of the deep venous system predisposes the patients to localized intravascular coagulopathy and thromboembolism with subsequent risk of pulmonary arterial hypertension.[5]
CLOVES Syndrome
CLOVES syndrome is characterized by congenital lipomatous overgrowth, vascular malformations, epidermal nevi, and spinal/skeletal anomalies. Capillary, lymphatic, venous malformations, or combined malformations are often seen in these patients. In rare cases, spinal arteriovenous malformations have also been described. The lipomatous masses are usually present over the trunk, and vascular malformations are often present within or overlying these masses. There can be a focal or segmental overgrowth of the extremities with predominant fibroadipose components. Scoliosis, macrodactyly, and sandal-gap deformity are some of the skeletal anomalies seen. Associated neurological impairment and visceral anomalies have also been described in some cases.[5]
Proteus Syndrome
Capillary, venous, or lymphatic malformation with progressive, severely deforming overgrowth of body parts, bony hypertrophy, dysregulated adipose tissue, and epidermal nevi are typically seen in Proteus syndrome. The hallmark feature of this syndrome is the cerebriform connective tissue thickening over the soles and palms.[6]
Diffuse Capillary Malformation with Overgrowth (DCMO)
Diffuse capillary malformation with overgrowth is a recently described clinical entity that falls in the spectrum of vascular anomalies with overgrowth. Patients can have a widespread reticulated capillary malformation affecting multiple contiguous sites, with a sharp midline demarcation associated with non-progressive soft tissue and/or bony hypertrophy.[7]
Capillary Malformation-Arteriovenous Malformation Syndrome (CM-AVM)
Capillary malformation-arteriovenous malformation syndrome is an autosomal dominant disorder caused by RASA1 mutation. A few cases with EPHB4 mutation have also been described. It is a combined vascular malformation characterized by multiple PWSs and arteriovenous malformation (AVM) or fistulas. Multiple PWSs that are multifocal, randomly distributed, pink-to-reddish brown colored, of variable size, with geographical margins, and with a pale/white halo are seen in the affected individual at birth or later in life. AVM may be present in the skin, muscle, bone, spine, or brain in one-third of these patients. Depending on the site, they can have various symptoms or life-threatening complications. Pain, bleeding, congestive heart failure, and neurological symptoms are some complications seen in CM-AVM.[4]
Parkes-Weber Syndrome
Parkes-Weber syndrome is characterized by multiple arteriovenous fistulas with bone and soft tissue hypertrophy, usually affecting the extremities. A RASA1 mutation is seen in patients having associated PWS; hence, it is considered a variant of CM-AVM.[16]
Macrocephaly-Capillary Malformation Syndrome (MCM)
Macrocephaly-capillary malformation syndrome is characterized by megalencephaly, brain and body asymmetry, capillary malformations, digital anomalies, and brain malformations. Often, PWS is seen on the central face, and patients can have seizures, developmental delay, and joint laxity.[5]
Phakomatosis Pigmentovascularis (PPV)
Phakomatosis pigmentovascularis is characterized by the coexistence of extensive capillary malformation and pigmented nevi. Five types of PPVs have been described based on the type of pigmented nevi (nevus spilus, dermal melanocytosis, epidermal nevus) and capillary malformation. Each type is further divided into 2 subtypes; subtype "a" denotes absence, and subtype "b" denotes the presence of extracutaneous findings. Patients can also have nevus anemicus and café-au-lait macules.
Nevus flammeus is a common feature in the first 4 types of PPV. It is associated with epidermal nevus type I, congenital dermal melanocytosis in type II, and nevus spilus in type III. Both congenital dermal melanocytosis and nevus spilus are seen in PPV type IV. Cutis marmorata telangiectasia congenita is associated with nevus spilus in PPV type V. Among the 5 types, PPV type II is most commonly seen.[6]
CLAPO Syndrome
CLAPO syndrome is a combination of Capillary vascular malformation of the lower lip, Lymphatic malformations of the head and neck, Asymmetry, and Partial or generalized Overgrowth. Capillary malformation of the lower lip, usually extending to the adjacent skin, lymphatic malformation of the tongue and neck, and asymmetric partial overgrowth of the face and extremities are the constellation of symptoms seen in this syndrome.[5]
Evaluation
The characteristic clinical feature helps in the easy diagnosis of PWS. The best timing of evaluation of a facial nevus flammeus is at birth.[2] It usually presents a diagnostic challenge when associated with other cutaneous or visceral vascular malformations and skin/soft tissue/visceral anomalies.
Ultrasound with color Doppler is useful in determining the flow resistance and thus identifying arteriovenous malformations (fast-flow vascular malformation) that clinically mimic PWS.[1][9] Computed tomography, magnetic resonance imaging, and magnetic resonance angiography can detect any superficial or deep tissue anomalies associated with PWS in the setting of a combined vascular malformation or a syndrome.[9] Imaging delineates the extent of these anomalies, assesses their severity, and ascertains the diagnosis.
A lesional biopsy is routinely not done to confirm the diagnosis; however, it can be used to differentiate from its clinical mimickers, prove the diagnosis of acquired PWS, and sample tissue for genetic studies.[5] Further evaluation may be required in cases where the PWS is suspected to be a part of a syndrome.
Treatment / Management
PWS is a vascular birthmark of a benign nature; therefore, reassuring the anxious parents may be the only management required. As the face is commonly affected, many find it cosmetically disfiguring, especially when associated with skin and soft tissue hypertrophy. Vascular-selective lasers are the mainstay of treatment, with the type of laser and laser parameters selected based on the color and thickness of PWS, the patient's skin type, prior treatment, and the response to it.[1][6][12]
Lasers work on the principle of selective photothermolysis. The laser energy gets preferentially absorbed by hemoglobin (target chromophore), which results in the heating and rupture of red blood cells and blood in the vasculature. Thus, laser energy gets converted into thermal energy, which diffuses to endothelial cells, causing selective damage to microvasculature through mechanical injury and photocoagulation.[12][17] The objective is complete photocoagulation of ectatic vessels in the lesional skin, which reduces dermal blood content and the lightening of PWS. Treatment initiation as early as infancy is found to have consistent improvement, better outcomes, and decreased risk of cutaneous hypertrophy and disfigurement. A deepening of color may occur, requiring a touch-up treatment later.[6][18]
A 585-nm short pulsed dye laser (PDL) is considered the gold standard in treating PWS.[1][6][12] During the initial treatment, the fluence used is 8 to 9 J/cm squared, with pulse duration and spot size varying between 0.45 ms to 40 ms and 2 mm to 12 mm, respectively.[17] Epidermal cooling allows safe and effective use of higher fluences and significantly reduces pain associated with the procedure. Different epidermal cooling methods exist, with cryogen spray cooling preferred over the others.[12] The clinical endpoint is a transient grey-blue discoloration of the treated area that evolves into purpura. Multiple treatment sessions may be required at intervals of 1 to 3 months.[17] Erythema, swelling, and bruising are the transient side effects commonly seen after laser treatment. Blistering and crusting are seen after overlapping pulses. The most serious complication from PDL is eye damage, especially when treating the periorbital area. Scarring, hyperpigmentation, and alopecia are less common but have more permanent adverse effects. Although more than 50% improvement will be seen in the majority, a complete clearance of the lesion may not be seen.[6][17] The response to treatment can vary depending on the clinical features listed below:[1][6][12]
- Anatomic site- Poor response is seen in PWS on acral and centrofacial sites.
- Size- More complete clearance is seen with smaller PWS.
- Color- Red PWS responds better than deep-purple colored.
- Thickening and nodularity- Longer wavelength lasers are more effective.
- Epidermal pigmentation- Energy gets absorbed by high melanin content in the epidermis, leading to reduced light penetration into the dermis.
In addition to the above-mentioned clinical attributes, neovasculogenesis occurring during the vascular remodeling phase post-procedure can also lead to unsatisfactory results.[12] In cases with suboptimal response, an increase in spot size, use of higher fluence, and overlapping of pulses may help. Although controversial and needing more evidence, the multiple-pass technique is theorized to increase the extent of vascular damage.
Longer wavelength lasers can penetrate deeper to damage the ectatic vessels located in the reticular dermis, thus justifying the use of alexandrite laser (755-nm) and neodymium-doped yttrium aluminum garnet (Nd:YAG) laser (1064-nm) in the treatment of darker/thicker/nodular lesions.[6][12] On the downside, they are associated with a greater risk of scarring and pigmentary changes. The dual sequential wavelength laser (DSWL), which combines PDL and Nd:YAG, emits 2 pulses of different wavelengths with a preselected delay from the same device. This targets vessels in both the superficial and deep dermis, using a lower fluence and thus producing a good response with fewer side effects. Frequency-doubled Nd:YAG laser, though useful, is found to be less efficacious. Intense pulsed light (IPL) with cutoff filters is an alternate treatment option with reduced efficacy but limited side effects.[12]
Photodynamic therapy (PDT) can be used alone or combined with lasers. PDT selectively targets hypervascular dermal lesions without causing epidermal damage and is comparable in efficacy or even superior to laser treatment. The treatment involves the intravenous administration of photosensitizer followed by irradiation of the affected area with light of appropriate wavelength, which produces reactive oxygen species that destroy the endothelial cells in the presence of oxygen.[6][17] Further studies are needed to optimize treatment protocols.
As angiogenesis during post-treatment vascular repair decreases the efficacy of laser therapy, angiogenesis inhibitors like imiquimod and rapamycin can be combined with lasers to improve the treatment response.[17] The use of local hypobaric pressure to increase the chances of photo-induced damage and site-specific pharmaco-laser therapy are some of the newer concepts in PWS treatment that require further studies to validate and optimize the treatment.[12]
Ablative lasers like carbon dioxide lasers can be used to treat hypertrophy and nodularity.[18] Surgical correction may be required in cases with soft tissue and bony overgrowth.
Cosmetic camouflage, skin grafting, cryotherapy, dermabrasion, tattooing, and radiation are some of the treatment modalities used in the past with poor cosmesis.[18]
Differential Diagnosis
The differential diagnosis of nevus flammeus includes the following:
- Salmon patch
- Early hemangioma
- Arteriovenous malformation
- Tufted angioma
- Eccrine angiomatous hamartoma
- Cutis marmorata telangiectatica congenita
- Segmental infantile hemangioma that may necessitate a workup for PHACES (Posterior fossa malformations, Hemangioma, Arterial anomalies, Coarctation of the aorta/cardiac defects, Eye abnormalities, and Sternal malformations)
- Nevus simplex
Prognosis
Isolated PWS is benign and almost always only of cosmetic concern.[13] Laser therapy at an early age gives better results as the progressive thickening and nodularity seen with age make it recalcitrant to treatment. A diagnosis needs to be established for cases where PWS is associated with other vascular malformations or a syndrome. This is crucial for early identification and management of skin complications and visceral anomalies and thus limits their impact on the patient's quality of life. The severity of associated anomalies determines the prognosis in these patients.
Complications
Glaucoma is a well-known complication of periocular PWS, with a greater risk seen with the involvement of eyelids.[17] Patients can develop progressive soft tissue and/or skeletal hyperplasia by the age of 50 years. Based on the site affected, there can be skeletal asymmetry and limb length discrepancies, which can be functionally disabling or cosmetically disfiguring.[6] The rare cases with mucosal involvement can have gingival hyperplasia predisposing to periodontal disease and poor dental hygiene.[1][9]
Pyogenic granuloma presents as a solitary nodule on a PWS that is often ulcerated and bleeds with trivial trauma. They are seen in adulthood and can develop spontaneously following trauma, oral contraceptive pills use, or during pregnancy. There have been isolated case reports of tufted angioma arising in a PWS.[3] There are documented cases of eczema arising solely in PWS or with a greater severity within PWS, which is analogous to the Meyerson phenomenon.[9] PWS, especially over the face, can be stigmatizing, leading to low self-esteem and mental stress. Several other complications and comorbidities can be seen with combined vascular malformations and syndromes.
Deterrence and Patient Education
Even though a red stain over a newborn's skin is often benign, it can be alarming to the parents. As important as it is to allay their fears, they need to be made aware of the natural evolution of the lesion and anticipated complications. Alert parents should be able to recognize any extracutaneous/systemic symptoms the child develops that allow the child to receive necessary health care early on.
Regular follow-up should be emphasized in cases like PWS over eyelids, combined vascular malformations, and a syndrome where the risk of complications is high. When indicated, the patient or their parents need to be informed of the treatment options available with their advantages and disadvantages, and they should be counseled regarding the need for early initiation of treatment to improve patient outcomes.
Enhancing Healthcare Team Outcomes
Enhancing patient-centered care, improving outcomes, ensuring patient safety, and enhancing team performance in managing nevus flammeus requires a collaborative approach involving physicians, advanced care practitioners, nurses, pharmacists, and other healthcare professionals. An interprofessional team may consist of a pediatric dermatologist, primary clinician, radiologist, vascular surgeon, pediatric surgeon, plastic surgeon, and orthopedic surgeon.
A child with PWS is usually first brought by the parents to a primary clinician who needs to differentiate it from its clinical mimickers. Health professionals should possess specialized skills related to nevus flammeus, including the ability to accurately diagnose the condition, choose appropriate treatment modalities (eg, laser therapy), and conduct regular assessments of the birthmark's progress. In a diagnostic dilemma, the child needs a referral to a dermatologist who can establish the diagnosis and recognize any possible syndromic association. If SWS is suspected, neurology and ophthalmology opinions should be sought. Even though PWS occurring alone is considered benign, it can also be rarely associated with complications. Skillful performance of laser procedures, if necessary, is essential to minimize complications.
Developing a comprehensive strategy for nevus flammeus care involves setting clear goals for patient outcomes, coordinating multidisciplinary care, and creating treatment plans tailored to each patient's unique needs and preferences. This strategy should consider factors such as the location and size of the birthmark, potential syndromic associations, and long-term management.
Coordinated care ensures that patients receive seamless, timely, and appropriate interventions. Healthcare professionals should collaborate on scheduling appointments, monitoring treatment progress, and addressing potential complications. Care coordination extends to providing emotional support to patients and families facing the psychosocial challenges associated with nevus flammeus.
By emphasizing these aspects of care, an interprofessional team can work together to provide patient-centered care for individuals with nevus flammeus. This approach enhances the likelihood of positive outcomes, ensures patient safety, and optimizes team performance, ultimately improving the overall quality of care for this condition.