Introduction
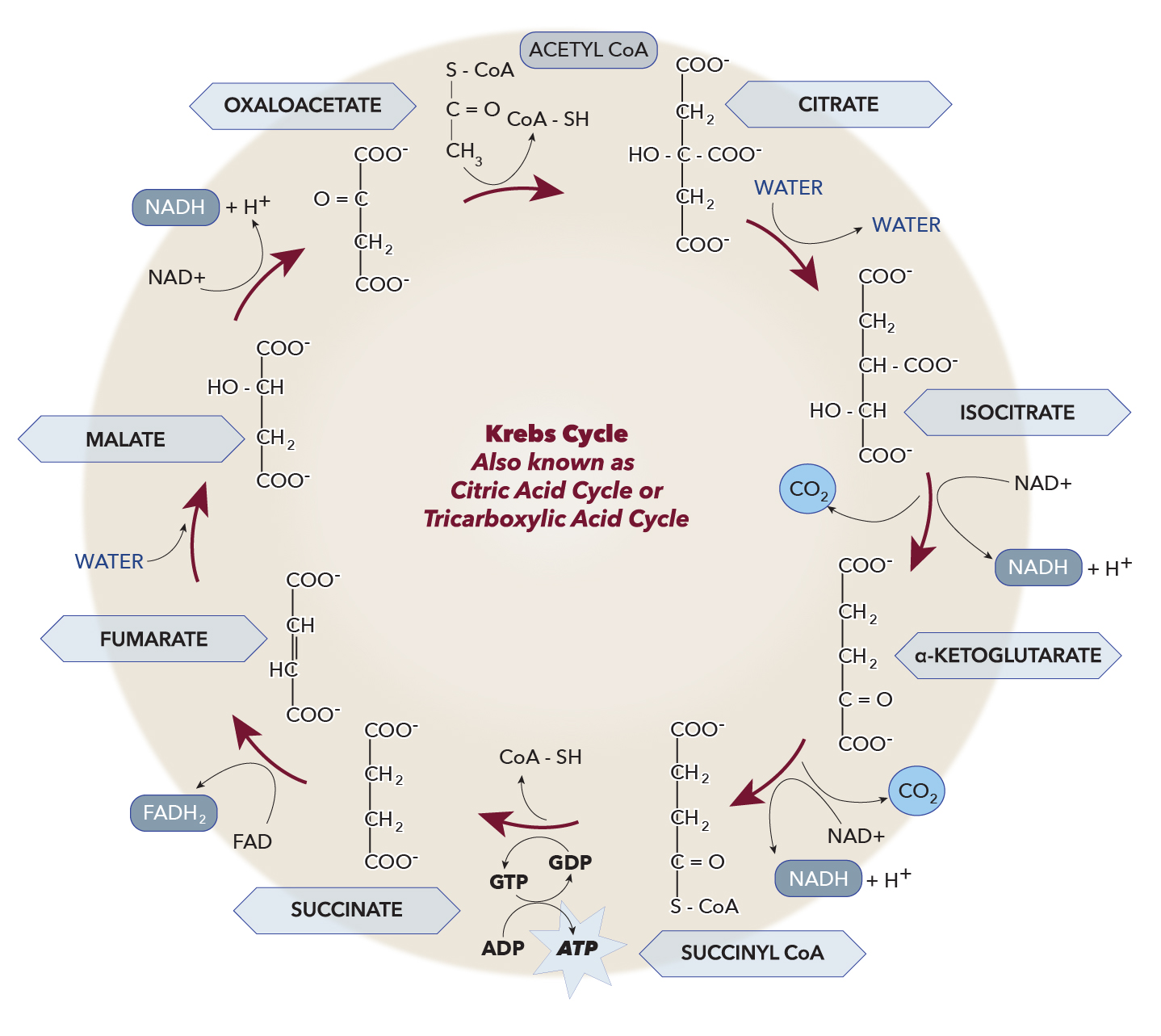
The tricarboxylic acid (TCA) cycle, also known as the Krebs cycle or citric acid cycle, is an important cell's metabolic hub (see Diagram. Krebs Cycle). It is composed of eight enzymes, all of which are within the mitochondrial matrix except the outlier succinate dehydrogenase, which is related to the respiratory chain on the inner mitochondrial membrane. The cycle serves as a gateway for aerobic metabolism for molecules that can convert to an acetyl group or dicarboxylic acid. Regulation of the TCA cycle occurs at three distinct points that include the three following enzymes: citrate synthase, isocitrate dehydrogenase, and alpha-ketoglutarate dehydrogenase. The cycle also plays a role in replenishing precursors for the storage form of fuels such as amino acids and cholesterol.[1]
Issues of Concern
The Krebs cycle, by itself, does not require the presence of oxygen; this element is necessary for the last stage of aerobic cellular respiration, i.e., oxidative phosphorylation.
Organic molecules endowed with energy (carbohydrates, lipids, proteins) are split in previous reactions. Before entering the Krebs cycle, they transform into acetyl-CoA, a molecule formed by an acetyl group (CH3CO-) and by an acyl transporter called coenzyme A.
However, the preferred source of acetyl-CoA remains glycolysis. The acetyl group is then oxidized, and the energy obtained is used for the synthesis of ATP in cooperation with oxidative phosphorylation. In eukaryotes, the Krebs cycle reactions take place in the mitochondrial matrix, a dense solution that surrounds the mitochondria crests: in addition to water, the matrix contains all the enzymes necessary for the biochemical reactions of the cycle, coenzymes, and phosphates.The Krebs cycle is controlled and regulated by the availability of the NAD+ and FAD substrates, while high concentrations of NADH inhibit it.
Cellular Level
Glucose metabolism takes place in the cytosol without the need for oxygen by a process termed glycolysis. It yields a small amount of ATP, and the three-carbon compound pyruvate. After the transportation of pyruvate into the mitochondria, pyruvate dehydrogenase complex (PDC) facilitates the conversion of pyruvate to acetyl-CoA and CO2. Each molecule of acetyl-CoA entering the TCA cycle yields 12 ATP molecules. The (PDC) has three protein subunits and requires five cofactors for its enzymatic function. The requirement of cofactors ensures the ability of the complex to be regulated. In high blood sugar levels, most acetyl-CoA will be derived from glucose, more specifically, pyruvate. However, in starvation or fasting states, beta-oxidation contributes to the production of acetyl-CoA. Acetyl-CoA undergoes oxidation to CO2 in eight steps, and the energy produced from these reactions is stored in NADH+H+, FADH2, and GTP. NADH+H+ and FADH2 then oxidize in the electron transport chain (mitochondrial respiratory chain), terminating in ATP synthesis.[2]
Intermediated from the TCA cycle are precursors for both catabolic and anabolic processes. It connects several metabolic processes (e.g., glycolysis, gluconeogenesis, ketogenesis, lipogenesis).[3]
These three mechanisms regulate the activity of pyruvate dehydrogenase complex (PDC): covalent modification being the primary form of regulation, allosteric regulation, and transcriptional regulation. The covalent modification takes place when the first subunit of the PDC, pyruvate decarboxylase, is phosphorylated. This phosphorylation results in decreased PDC activity and an increase of ADP or pyruvate (signaling the requirement for more acetyl-CoA in the TCA cycle, which downregulates PDC). Calcium ions upregulate phosphatase’s activity; phosphatase, in turn, dephosphorylates PDC rendering it active. Allosteric regulation of PDC entails the direct mechanism of substrate activation or product inhibition. For instance, when there is an excess release of Acetyl-CoA from E2 or NADH from E3, these products act to inhibit PDC directly. In contrast, an increase in the levels of CoASH (precursor to acetyl-CoA) or NAD+ will directly activate PDC. The last type of regulation of PDC activity is transcriptional regulation, which is dependent on the number of enzymes produced in fasting and fed conditions. In the fed state, enzyme production increases due to the effect of insulin, but it reduces in fasting states.[4]
Development
The Krebs cycle is also crucial during development. To make an example, the energy obtained from this metabolic pathway is essential for the adequate growth of the endothelial system, which will guide the formation of the blood and lymphatic vessels.
If the different phases of the Krebs cycle are not present in the fetal period, the baby's heart may have problems at birth. The alteration of the cycle causes an increase in cortisol, which alters the metabolism of the placenta and fetal development, including the correct function of the heart of the future child. These alterations can lead to death.
Organ Systems Involved
The Krebs cycle is present in every cell that uses oxygen to produce energy. This metabolic pathway is used as an anabolic cellular principle but also in the presence of catabolism.
Function
Citrate Synthesis
The enzyme citrate synthase catalyzes the formation of citrate from acetyl CoA and oxaloacetate, often regarded as the first step of the TCA cycle. This reaction is virtually irreversible and has a delta-G-prime of -7.7 Kcal/M that strongly favors citrate formation. The availability of substrates and products regulates the activity of citrate synthase. For instance, citrate itself acts as an inhibitor for citrate synthase, while oxaloacetate’s binding to it increases its affinity for acetyl-CoA. It bears mention that phosphofructokinase-1 in glycolysis is inhibited by citrate, while it activates acetyl-CoA carboxylase for fatty acid synthesis. This point illustrates the interconnectivity of our metabolic cycles.[5]
Isomerization of Citrate
The reversible conversion of citrate to isocitrate is catalyzed by the enzyme aconitase, which contains an iron-sulfur center that facilitates the hydroxyl group migration. Cis-aconitate is the intermediate product of this reaction.[6]
Oxidative Decarboxylation of Isocitrate
The oxidative decarboxylation of isocitrate to alpha-ketoglutarate becomes catalyzed by NAD+ -dependent isocitrate dehydrogenase producing CO2, NADH, and a proton; this is the rate-limiting step of the TCA cycle. Production of the first reduced coenzyme in the cycle takes place at this reaction. The tendency of this reaction to produce gas makes it irreversible. ADP and calcium ions allosterically regulate isocitrate dehydrogenase by activating it, while ATP and NADH inhibit its activity.[7]
Oxidative Decarboxylation of Alpha-ketoglutarate
The conversion of alpha-ketoglutarate to succinyl-CoA is catalyzed by the alpha-ketoglutarate dehydrogenase complex producing NADH, CO2, and H+. The function of the alpha-ketoglutarate dehydrogenase complex is analogous to PDC. Alpha-ketoglutarate becomes decarboxylated by E1 of this complex, transferring the four remaining carbons to thiamine pyrophosphate. Thiamine pyrophosphate is the first cofactor. Then the succinyl group transfers to CoASH by E2 with the help of FAD. The final step involves the resynthesis of FAD along with NADH from NAD+ by E3. This last step ensures the maintenance of substrates and cofactors required to continue the dehydrogenase complex activity. The cofactors necessary for alpha-ketoglutarate dehydrogenase complex include thiamine pyrophosphate, lipoic acid, coenzyme A, NAD+, and FAD. Succinyl-CoA, NADH, and ATP inhibit the alpha-ketoglutarate dehydrogenase complex.[8][9]
Cleavage of Succinyl Coenzyme A
The enzyme succinate thiokinase catalyzes the reversible interconversion of succinyl-CoA to succinate by cleaving succinyl CoA’s thioester bond. The enzyme uses inorganic phosphate and dinucleotide to produce trinucleotide and CoA. This coupled reaction is called substrate-level phosphorylation, just like what happens in glycolysis.[10]
Oxidation of Succinate
Succinate dehydrogenase is also called complex II due to its role in the aerobic respiration chain. It catalyzes the reduction of ubiquinone to ubiquinol. The TCA cycle catalyzes the oxidation of succinate to fumarate, producing a reduced FADH2 from FAD.[11]
Hydration of Fumarate
The reversible hydration of fumarate to malate is catalyzed by fumarase (or fumarate hydratase). In another attempt to illustrate the interconnectedness of metabolic pathways, note that fumarate production also occurs in the urea cycle.[12]
Oxidation of Malate
Malate dehydrogenase is the catalyst in the reversible oxidation of malate to oxaloacetate, which is the last step of the TCA cycle. This enzyme plays a crucial role in NADH oxidation within the TCA cycle. The delta-G-prime is positive, which indicates that the reaction favors malate. However, the consumption of oxaloacetate by citrate synthase drives this reaction forward to produce more oxaloacetate.[13]
Cataplerotic Processes
Citric acid intermediates can leave the cycle to participate in the biosynthesis of other compounds. Citrate can be directed toward fatty acids; succinyl-CoA to heme synthesis; alpha-ketoglutarate to amino acid synthesis, purine synthesis, and neurotransmitter synthesis; oxaloacetate to amino acid synthesis, and malate to gluconeogenesis.[14][4]
Anaplerotic Processes
Intermediates are inserted into the TCA cycle to replace the cataplerotic processes and ensure the continuation of the cycle. For instance, pyruvate can enter the cycle throughout the body through pyruvate carboxylase, which inserts additional oxaloacetate into the cycle. This process causes the reaction to be pushed forward toward the already exergonic citrate synthase, as there is more oxaloacetate to participate in the reaction. The liver is another example, as it can produce alpha-ketoglutarate by oxidative deamination or transamination of glutamate.[15][4]
Related Testing
Evaluating mitochondrial function involves evaluating the Krebs cycle. For example, in nonalcoholic liver disease (NAFLD), there is a mitochondrial malfunction, and it is one of the diagnostic cornerstones. Some authors suggest comparing the plasma values of isocitrate and citrate for a finding of mitochondrial alteration.
The search for plasma values of mitochondrial metabolites can be used to understand how mitochondria are working.
Pathophysiology
Mitochondrial dysfunction can result from an excess of calories introduced through food; the Krebs cycle can no longer find a balance between the molecules to be degraded and the number of molecules available. Obesity shows a mitochondrial alteration, with an increase in oxidative stress and the production of reactive oxygen species, inflammation, and apoptosis.
Mitochondrial dysfunction can also mean overwork compared to normal values. In Duchenne pathology (in an animal model), an increase of mitochondrial metabolites is present in different tissues, such as the diaphragm and peripheral muscles, the central nervous system. The reasons are probably related to oxidative stress.
Clinical Significance
Pyruvate Dehydrogenase Complex Deficiency
Pyruvate dehydrogenase complex deficiency is a neurodegenerative disorder due to an abnormal pyruvate decarboxylase subunit caused by a mutated X-linked PDHAD gene. This mutation leads to the impaired conversion of pyruvate to acetyl-CoA. Since there is an excess accumulation of pyruvate, lactate dehydrogenase will convert it to lactate leading to potentially fatal metabolic acidosis. Other symptoms include neonatal-onset lethargy, hypotonicity, muscle spasticity, neurodegeneration, and early death.[16][17][18]
Leigh Syndrome
Subacute necrotizing encephalomyelopathy or Leigh syndrome is a progressive neurological disorder due to gene mutations encoding proteins of the PDC. In most children, the first observed sign is the inability to perform motor skills previously acquired. Other symptoms include loss of head control, poor suckling, recurrent vomiting, and loss of appetite.[19][20][21]
Thiamine Deficiency
Thiamine deficiency is similar to pyruvate dehydrogenase complex deficiency in that it will lead to the shunting of pyruvate to lactate leading to metabolic acidosis. However, the culprit here is a deficiency in the active form of thiamine (thiamine pyrophosphate) rather than PDC. Acute thiamine deficiency is dry beriberi, while chronic thiamine deficiency is wet beriberi. Dry beriberi characteristically demonstrates diminished reflexes and symmetric peripheral neuropathy with motor and sensory changes. On the other hand, wet beriberi classically affects the heart leading to tachycardia, dilated cardiomyopathy, high-output congestive heart failure, and peripheral edema.[22][23]
Fumarase Deficiency
Fumarase deficiency is a rare autosomal recessive metabolic disorder of the TCA cycle due to a mutation in the FH gene. It is characterized by a deficiency of the enzyme fumarase hydrates, which leads to the buildup of fumaric acid. It is a condition that mainly affects the nervous system. Affected children may have severe developmental delay, microcephaly, hypotonia, encephalopathy, seizures, psychomotor retardation, and failure to thrive.[24][25][26][27]
Mutations of Isocitrate Dehydrogenase
Researchers have found mutations of isocitrate dehydrogenase in several types of cancers, including leukemia, gliomas, and sarcomas. IDH mutations can be useful for the differential diagnosis and subclassification of human gliomas. The normal function of IDH is to catalyze the oxidative decarboxylation of isocitrate to alpha-ketoglutarate. Mutant IDH catalyzes the formation of 2-hydroxyglutarate instead of alpha-ketoglutarate. 2-hydroxyglutarate is an oncometabolite that causes DNA and histone hypermethylation leading to neoplasia. 2-hydroxyglutarate can be used as a biomarker for cancer in patients with inborn errors of metabolism.[28][29][7][30]