Introduction
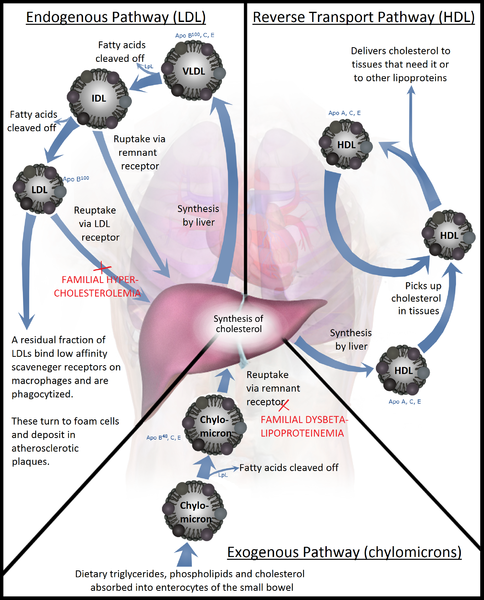
Lipoproteins are complex particles that transport lipids, such as phospholipids, triglycerides, and cholesterol, between cells. There are 5 different types of lipoproteins, which are classified according to their density and composition: chylomicrons, very-low-density lipoprotein (VLDL), intermediate-density lipoprotein (IDL), low-density lipoprotein (LDL), and high-density lipoprotein (HDL). As per its denomination, HDL has the highest density of lipoproteins and the highest proportion of proteins to lipids.[1] HDL is particularly interesting in medicine, as research has shown a strong inverse association between HDL cholesterol concentration and the risk of atherosclerosis (see Image. Lipoprotein Metabolism).[2]
Molecular Level
HDL is composed of cholesterol, triglycerides, and various apolipoproteins, including:
- Apo-AI: The primary structural apolipoprotein of HDL; activates Lecithin–Cholesterol AcylTransferase (LCAT)
- Apo-AII: A structural protein in HDL; acts as an activator of hepatic lipase
- Apo-IV: Has an unknown function
- Apo-AV: Activates lipoprotein lipase (LPL), responsible for triglyceride lipolysis
- Apo-CI: Responsible for activating LCAT
- Apo-CII: Responsible for activating LPL
- Apo-CIII: Responsible for inhibiting LPL
- Apo-E: Ligand for the LDL receptor
Function
The primary function of HDL is the transport of cholesterol from the peripheral tissues to the liver, playing a role in the biodistribution of lipids.[3] HDL is known for its antiatherogenic and antiinflammatory properties, thanks to its uptake and return of the cholesterol stored in the foam cells of atherosclerotic plaques to the liver. Thus reducing the size of the plaque and its associated inflammation.[4]
Mechanism
HDL synthesis occurs in the liver and the intestines. The first step of HDL synthesis involves the production of its main structural apolipoproteins, Apo-AI. Apo-AI is the structural protein of HDL and receives cholesterol and phospholipids from enterocytes and hepatocytes via transporter ABCA1, forming pre-beta HDL. HDL travels in circulation and receives free cholesterol and phospholipids from peripheral tissues, chylomicrons, and VLDL. As mentioned, Apo-AI acts as a cofactor for LCAT. LCAT converts free cholesterol at the surface of HDL to cholesterol esters, which are then incorporated into the cholesterol ester core of HDL.[1]
Cells in peripheral tissues are exposed to and accumulate cholesterol through the circulation and de novo synthesis. However, since most cells can not catabolize cholesterol, they require a reverse transport mechanism to return cholesterol to the liver. ABCG1 transfers cholesterol in peripheral cells to HDL, like ABCA1 in the liver, and allows the transfer of cholesterol to HDL in the circulation. Subsequently, LCAT incorporates this free cholesterol into the HDL particle, eventually leading to its uptake in the liver through 3 distinct pathways: the cholesterol ester transfer protein (CETP) pathway, the LDL receptor pathway, and the Scavenger receptor class B type 1 (SR-B1) pathway.[5][6]
The CETP pathway involves the transfer of lipids between HDL and other lipoproteins. For instance, CETP exchanges the cholesterol esters in the core of HDL particles to Apo-B-containing particles, VLDL, and LDL in exchange for triglycerides. The cholesterol, now contained in VLDL and LDL, can undergo uptake by the LDL receptor, resulting in the return of cholesterol from peripheral tissues to the liver. This exchange also enriches HDL in triglycerides and depletes the cholesterol ester core. As a result, hepatic lipases can metabolize the triglyceride-rich HDL, resulting in small HDL particles. These small HDL particles release their Apo-AI. The liver can then degrade Apo-A1 through an unknown mechanism, or the kidney can clear the free Apo-A1. It is also possible that HDL particles contain Apo-E, which allows HDL particles to undergo uptake by the LDL receptors into the liver and subsequently degrade. Endothelial cell lipase also metabolizes Apo-AI, leading to its clearance from circulation. In brief, the CETP pathway results in both the transfer of cholesterol in HDL to the liver and the elimination of HDL from the circulation through the degradation of Apo-AI.[6][7]
The liver can also uptake HDL via its apolipoprotein Apo-E. Apo-E interacts with the LDL receptor and leads to the uptake of the entire LDL particle. The SR-B1 pathway is responsible for the uptake of HDL cholesterol into the liver without the internalization of the HDL particle. After cholesterol uptake, the smaller HDL particle gets released back into the circulation. SR-B1 deficiency results in increased HDL levels but correlates with an increased, not decreased, risk of atherosclerosis.[8]
Testing
HDL is tested through blood sampling and commonly determined within a complete lipid profile. Testing in adults requires fasting 12 hours before sampling, whereas testing is possible without fasting in the youth without risk factors for heart disease. Without fasting, only total cholesterol and HDL are usable. In this scenario, if the total cholesterol is 200 mg/dL or above or HDL is below 40 mg/dL, the NCEP III guidelines indicate a follow-up lipoprotein profile is necessary for management based on LDL.[9]
A lipid profile, and thus HDL-C, may be ordered for those with risk factors for heart disease, which includes:
- Smoking (cigarettes)
- Sedentary lifestyle
- Being overweight or obesity
- Poor diet
- Men over 45 years or women older than 55 years
- Hypertension (>140/90)
- Positive immediate family history of premature heart disease (under 55 for men and under 65 for women)
- Pre-existing heart condition
- Diabetes or prediabetes
For youth, the American Academy of Pediatrics recommends that children be tested once between ages 9 and 11 and repeated between the ages of 17 and 21. This screening is recommended for children under the age of 9 if their parents test positive for high cholesterol. The NCEP ATP III guidelines also suggest using agents that correct the HDL abnormality while also lowering LDL cholesterol, lowering LDL cholesterol being paramount.
Monitoring the lipid profile involves testing to be completed 4 to 12 weeks after starting a statin and every 3 to 12 months after that to ensure a response. These levels can also be checked after successful lifestyle changes, such as smoking cessation, diet, and exercise. Research has shown that a higher intake of total fat in the form of unsaturated fat can reduce triglycerides and raise HDL cholesterol in metabolic syndrome.
The ideal lipid profile is controversial. The NCEP ATP III guidelines indicate that the ideal HDL levels should be between 40 and 60 mg/dL. Some laboratory reports give a ratio of total cholesterol to HDL cholesterol, with a desirable ratio of 5 to 1 and an optimum ratio of 3.5 to 1. It bears mentioning that lipid profiles should not be measured when a person is acutely ill or is less than 6 weeks recovering from an illness, as cholesterol t is lowered during acute illness, stress (surgery or trauma), or pregnancy.
Pathophysiology
The ability of HDL to uptake and return excess cholesterol from peripheral tissues back to the liver is of particular interest because of its role in preventing atherosclerosis, the precursor to myocardial infarction, transient ischemic attack, and stroke. Atherosclerosis generally begins in areas of nonlaminar flow, such as branch points and the inner curvature of arteries. This nonlaminar flow increases the shear stress of the artery wall and results in inflammation and activation of the NF-κB pathway. The NF-κB pathway leads to the expression of adhesin molecules, which recruits macrophages to the intima of the artery wall. Simultaneously, the turbulent flow of the blood in the arteries also leads to the disruption of the tight junctions between the endothelium of the artery, resulting in the deposition of LDL and macrophages. In the intima of the artery, the LDL particles undergo oxidation, leading to the macrophages' prolonged activation in the intima and further cascades of inflammation. These macrophages bind and uptake the oxidized LDL, process the LDL cholesterol, and store the cholesterol as cytoplasmic droplets. These cholesterol-filled macrophages have a foamy appearance and are called foam cells. Unfortunately, these foam cells remain in the intima of the artery and continue to promote inflammation, increasing the size of the atherosclerotic plaque. Eventually, these atherosclerotic plaques close the vessel's lumen, leading to angina symptoms. When the plaques eventually rupture, they lead to thrombosis and subsequent occlusion of the artery lumen, resulting in the previously mentioned cardiovascular events.[10][11][12]
HDL is of interest in atherosclerosis as it can extract cholesterol from foam cells and lead to the remission of atherosclerotic plaque. Specifically, the free cholesterol synthesized in the foam cells is effluxed by ABCA1, SR-B1, and ABCG1 into already circulating HDL or onto Apo-AI or Apo-E discs, forming HDL. In this manner, HDL is considered an antiinflammatory, as it removes free cholesterol from foam cells and results in the down-regulation of inflammation within the atherosclerotic plaque. However, it bears mentioning that HDL levels do not always correlate with decreased risk of atherosclerosis, as HDL cholesterol levels can increase in cases where the reverse cholesterol transport pathway is reduced, such as SR-B1 deficiency.[5]
Clinical Significance
Hypoalphalipoproteinemia is a deficiency of HDL. These diseases correlate with a mild increase in triglycerides and are inherited in an autosomal recessive pattern. Defects in the lipid pathways that lead to hypoalphalipoproteinemia included problems with APOA1, ABCA1 (Tangier disease), and LCAT (Fish Eye Disease). In Tangier disease, functional mutations in ABCA1 reduce the ability of ABCA1 to lipidate newly secreted Apo-AI in the liver and intestines, leading to rapid catabolism and clearance of Apo-A1 and low HDL levels. In mice, an 80% reduction in HDL occurs with ABCA1 knockout in the liver, and a 30% reduction in HDL occurs with ABCA1 knockout in the intestines. In Fish Eye Disease, LCAT deficiency reduces HDL cholesterol and Apo-AI levels, resulting in a higher percentage of small HDL particles but an overall reduction in HDL. Humans have high HDL cholesterol levels and large HDL particles in CETP deficiency. Here, CETP cannot exchange cholesterol for triglycerides between HDL VLDL and LDL, leading to the reduced breakdown of HDL and increased HDL size.[13][14]
Elevated HDL levels are associated with a decreased risk of coronary heart disease and are thought to play a role in predicting cardiovascular risk. However, cholesterol ester transfer protein deficiency, primary familial hyperalphalipoproteinemia, and endothelial lipase deficiency result in extremely high levels of HDL, which may mitigate the protective effects and even, paradoxically, increase cardiovascular risk.[15] Additionally, there are secondary causes that can cause high levels of HDL to accumulate, such as primary biliary cirrhosis and thyroid disorders.[16][17] HDL levels can also increase with lifestyle changes, such as smoking cessation, diet, and exercise.[9]
Studies are ongoing on the relationship between HDL and cardiovascular risk, as mixed reports have been made. One study reported a 22% decrease in cardiovascular risk with each 15 mg/dl increase in HDL. However, CETP inhibitors, which can increase HDL levels by 100%, lead to a 60% increase in cardiovascular events.[18]