Continuing Education Activity
Idiopathic inflammatory myositis (IIM) is classified into four subtypes based on clinical and histopathological features. Polymyositis (PM) and dermatomyositis (DM) are 2 of those subtypes in addition to immune-mediated necrotizing myopathy and inclusion body myositis. The presence of autoantibodies and inflammatory infiltration in the muscles suggests that idiopathic inflammatory myositis is an autoimmune condition, but the target autoantigens have not yet been identified. Apart from symmetric proximal muscle weakness, polymyositis and dermatomyositis have several clinical manifestations in common when compared to inclusion body myositis and necrotizing myopathy and can be grouped for discussion. The most commonly used criteria for PM and DM are Peter/Bohan criteria: symmetric proximal muscle weakness, elevated serum muscle enzymes, myopathic changes in electromyography (EMG); Characteristic muscle biopsy abnormalities; and the typical rash of dermatomyositis. This activity reviews the evaluation and management of dermatomyositis and polymyositis and the role of interprofessional team members in collaborating to provide well-coordinated care and enhance patient outcomes.
Objectives:
- Review the presentation of polymyositis and dermatomyositis.
- Describe the evaluation of patients with polymyositis and dermatomyositis.
- Summarize the treatment of polymyositis and dermatomyositis.
- Outline the evaluation and management of dermatomyositis and polymyositis and the role of interprofessional team members in collaborating to provide well-coordinated care and enhance patient outcomes.
Introduction
Idiopathic inflammatory myositis (IIM) is classified into four subtypes based on clinical and histopathological features. Polymyositis and dermatomyositis (PM/DM) are two of those subtypes, in addition to immune-mediated necrotizing myopathy and inclusion body myositis. The presence of autoantibodies and inflammatory infiltration in the muscles suggests that idiopathic inflammatory myositis is an autoimmune condition, but the target autoantigens have not yet been identified. Apart from symmetric proximal muscle weakness, polymyositis and dermatomyositis (DM) have several clinical manifestations in common when compared to inclusion body myositis and necrotizing myopathy and can be grouped for discussion.
The most commonly used criteria for PM/DM are Peter/Bohan criteria.[1]
- Symmetric proximal muscle weakness
- Elevated serum muscle enzymes
- Myopathic changes in electromyography (EMG)
- Characteristic muscle biopsy abnormalities
- Typical rash of dermatomyositis
Etiology
Inflammatory myopathies are immune-mediated processes triggered by environmental factors in genetically susceptible people.
- Environmental factors: The best-known environmental risk factors are drugs, infections, ultraviolet (UV) light, vitamin D deficiency, and smoking. Few cases of myositis resembling polymyositis and dermatomyositis (PM/DM) have been reported after treatment with penicillamine, interferon (IFN)-alpha, and anti-tumor necrosis factor (TNF) inhibitors. The detection of antibodies to the Coxsackie virus, more frequently in patients with juvenile diabetes mellitus than in controls, supports the hypothesis of an infectious trigger. However, solid evidence is lacking for any role of continuing viral infection as a cause of chronic muscle inflammation.
- Genetic susceptibility: The occurrence of myositis in monozygotic twins and first-degree relatives of affected individuals suggests a role for genetic factors in developing IIM. The genetic factors are complex because the human leukocyte antigen (HLA) genes associated with IIM vary in different populations worldwide. The strongest known risk factors are HLA DRB1 *0301 and the linked DQA1 *0501.
Epidemiology
The estimated prevalence of polymyositis and dermatomyositis (PM/DM) is 5 to 22 per 100,000 persons, and the incidence is approximately 1.2 to 19 per million persons at risk per year. The incidence of myositis is increasing over time due to an increase in the detection rate. Dermatomyositis is bimodal in age distribution, with one peak at 5 to 15 years and another at 45 to 60 years. Polymyositis rarely occurs in the pediatric age group, and the mean age is between 50 and 60 years. The overall female-to-male incidence ratio is 2 to 3:1. In the United States, the Black race to White race ratio of incidence is 3 to 4:1. In Europe, the prevalence significantly increases from the north to the south, and this may be due to either environmental or genetic reasons.
Pathophysiology
The following are the proposed pathogenetic mechanisms:
- The direct effect of inflammatory cell infiltrates includes CD4+ and CD8+ T cells, B Cells, macrophages, and dendritic cells.
- Indirect effects of cytokines include interleukins, tumor necrosis factors, and interferons.
- Involvement of microvasculature includes increased thickening of endothelial cells, both of capillaries and venules, which are noted in myositis. This, along with increased expression of adhesion molecules, can promote inflammatory cell infiltration of the muscle tissue. Clinical observation of nail splinters and histological observation of decreased capillaries in muscle tissue supports this pathogenic mechanism.
- A humoral mechanism is suggested based on the presence of autoantibodies in serum and complement and immunoglobulin in the muscle biopsy specimen.
Histopathology
The characteristic histopathologic features of polymyositis and dermatomyositis (PM/DM) are the presence of mononuclear inflammatory cell infiltrates and degeneration and regeneration of muscle fibers. Atrophy of muscle fibers in a perifascicular fashion is the hallmark feature of dermatomyositis.
Immunopathology: Endomysial inflammatory cell infiltrates are mainly composed of CD8+T cells, and in addition, CD4+T cells, macrophages, and dendritic cells are seen in polymyositis. Perivascular and perimysial infiltration of CD4+T cells, macrophages, dendritic cells, and B cells are seen in dermatomyositis. The difference in immunohistopathologic features of these two subsets of myositis suggests that different pathogenic mechanisms could be involved in them, with T-cell-mediated muscle damage in PM and a role for microvessels in DM.
Increased expression of MHC class I and II molecules are present both on regenerating fibers and normal-appearing muscle fibers in patients with myositis. The MHC Class I antigen expression is more frequently observed than MHC class II, and the mechanism by which the MHC expression in muscle is induced has not been elucidated.
History and Physical
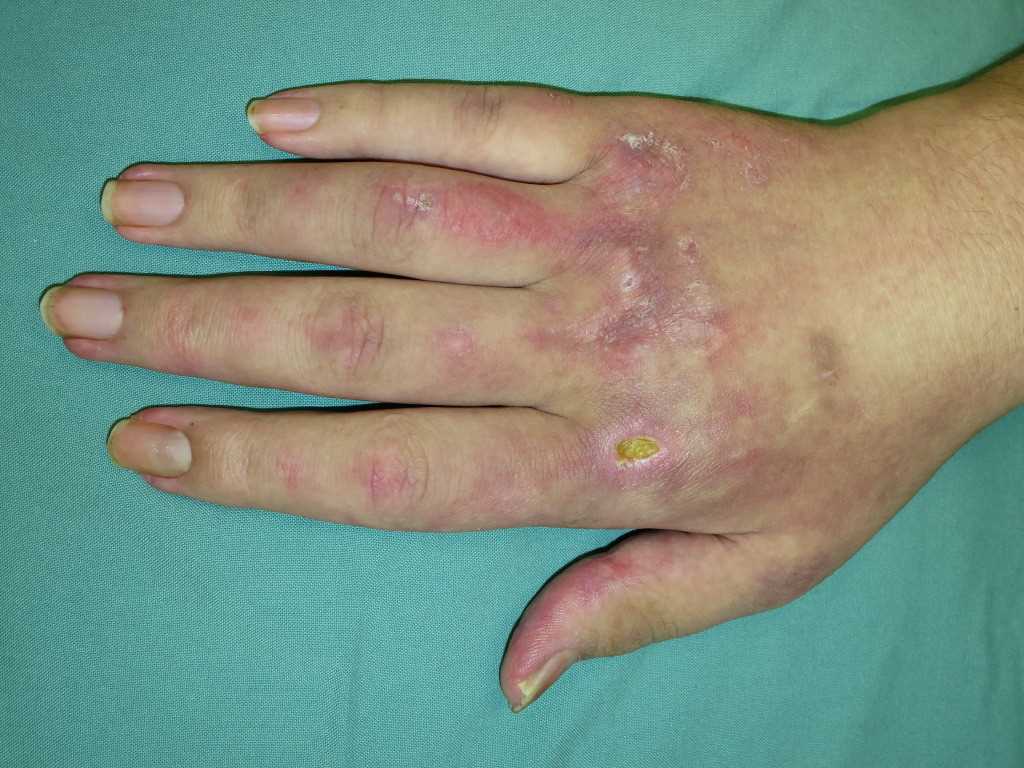
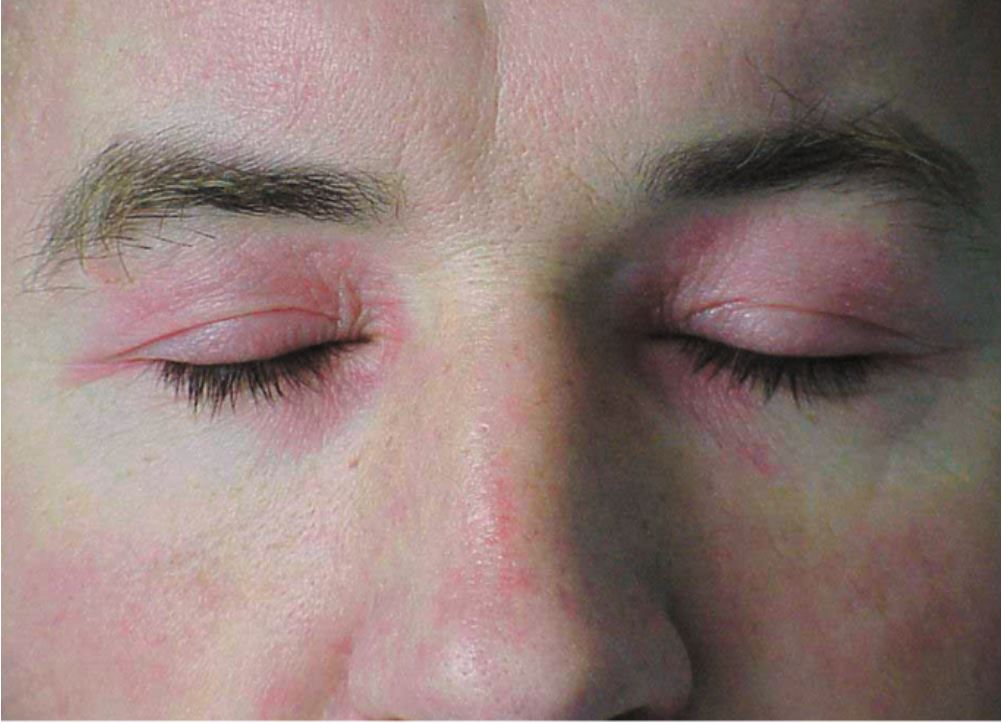
The hallmark clinical feature of IIM is symmetric proximal muscle weakness. It is manifested as difficulty in getting up from a sitting position, climbing stairs, lifting heavy objects, and overhead abduction of the arm.In addition to muscle weakness, patients with dermatomyositis often present with a characteristic skin rash. Gottron papule and heliotrope rash are the pathognomonic skin finding of dermatomyositis.
A description of the major skin findings in dermatomyositis is given below:
- Heliotrope rash: Violaceous rash on the eyelids sometimes associated with periorbital edema
- Gottron papules: Erythematous rash with variable papules on the extensor aspect of digits (metacarpophalangeal, proximal interphalangeal, and distal interphalangeal joints)
- Gottron sign: Erythematous papules, macules, or patches on sites other than hands, particularly extensor surfaces of elbows, knees, and ankles.
- Shawl sign: Erythematous rash on the anterior chest (in a V-sign) and upper back
- Mechanic’s hands: Dilated capillary loops at the base of the fingernails, irregular and thickened cuticles, and cracked palmar fingertips
- Holster sign: Poikiloderma on the lateral aspects of the thigh (areas protected from sunlight)
These lesions (except the holster sign) are photosensitive and may be aggravated by ultraviolet radiation. Other manifestations include dystrophic cuticles, peri-ungual erythema, abnormal nail bed capillary loops, diffuse flat erythema over the forehead, chin, and knees; photosensitivity; shawl sign; V-sign; panniculitis; and calcinosis cutis.
Dermatomyositis includes several clinically distinct phenotypes, and specific autoantibodies are associated with each unique phenotype.
Patients with dermatomyositis have little or no muscle involvement and are referred to as hypomyopathic or amyopathic dermatomyositis.
Extramuscular manifestations of IIM include arthritis, Raynaud phenomenon, dysphagia, and interstitial lung disease.
Evaluation
When a patient's history and proximal weakness are compatible, they should initially be evaluated by measuring the muscle enzymes, including creatine phosphokinase (CPK), aspartate aminotransferase, alanine aminotransferase, lactate dehydrogenase (LDH), and aldolase. CPK level usually correlates with the severity of muscle inflammation but, rarely, can be normal. Serial muscle enzyme measurements are useful for following myositis activity and are used to supplement serial strength testing. Inflammatory markers like ESR and CRP can also be elevated. Autoantibodies, muscle imaging, electrophysiologic examination, and muscle biopsy can be used to confirm the diagnosis. Autoantibodies are present in more than 80% of patients with inflammatory myositis. They are classified as (1) myositis-specific antibodies and (2) myositis-associated antibodies.
- Myositis-specific antibodies (MSA) have a specificity of 90% and are associated with distinct phenotypes. They include antisynthetases and dermatitis-associated antibodies. Antisynthetases are a group of antibodies that target the cytoplasmic aminoacyl-tRNA synthetase enzymes(ARS). Anti Jo-1 is the most studied and commonly detected antisynthetase antibody. Anti-Mi-2, anti-MDA5, anti-TIF, anti-SAE, and anti-NXP are dermatomyositis-associated antibodies.[2]
- Myositis-associated antibodies (MAA) are not disease-specific and are frequently seen in patients with overlap syndrome. The myositis-associated antibodies are anti-PM SCL, anti-Ro, anti-La, anti-Ku, and anti-U1 RNP.
If the muscle enzymes are abnormal, an objective evaluation of muscle inflammation is performed with EMG or MRI to document muscle abnormality and to identify a site for biopsy.
Abnormal findings expected in EMG are low amplitude, short duration, and polyphasic potential; increased membrane irritability; spontaneous fibrillation; and early recruitment. EMG should be done only on one side to reserve the contralateral side for the muscle biopsy to avoid electrical burn artifacts. MRI using short tau inversion recovery (STIR) sequence detects focal inflammation as bright signals, and a T1 sequence is useful in detecting atrophy and scarring.
Muscle biopsy helps differentiate polymyositis and dermatomyositis (PM/DM) based on their distinct histopathological features. In patients with dermatomyositis, a skin biopsy will show mild atrophy of the epidermis with vascular changes in the basal layer and perivascular inflammatory infiltrates in the dermis.
A chest X-ray should be done in all patients with PM or DM to detect lung involvement, such as interstitial lung disease. Pulmonary function tests and CT chest can be done based on the patient's symptoms and in cases of an abnormal chest X-ray. An increased prevalence of malignancy is described in patients with inflammatory myositis, particularly dermatomyositis.[3] Hence, age-appropriate screening with colonoscopy, low dose CT chest, Pap smear, and mammogram, in addition to the routine blood count, chemistries, and urinalysis, must be done in these patients. Patients with dermatomyositis who have positive anti-NXP2 or anti-TIF one antibody are at increased risk of cancer within three years of their diagnosis; therefore, PET/CT scans should also be considered.
Treatment / Management
Corticosteroids are the initial therapy and cornerstone in managing both polymyositis and dermatomyositis (PM/DM).[4] The common practice is to start with prednisone 1 to 2 mg/kg/day. In patients with severe weakness and those with extramuscular involvement, intravenous methylprednisolone 1mg/day for three consecutive days may be used. Patients should be closely followed, and a steroid dose can be slowly tapered based on the clinical response. There is no guideline for the initial dosing or tapering of steroids, so the treatment should be under an experienced physician and based on clinical judgment. Patients and families should be educated about the disease and the treatment. Patients should be monitored for steroid-related adverse effects, particularly osteoporosis, steroid-induced myopathy, hypertension, hyperglycemia, and weight gain.
Steroid-sparing therapy like methotrexate and azathioprine can be used in patients who develop adverse effects to steroids or those having trouble with tapering the steroids in patients with normal thiopurine methyltransferase (TPMT) activity. They are also used in patients with inadequate responses to steroids. Methotrexate is effective in treating muscle and joint symptoms but should be used cautiously in patients with lung involvement because of its potential lung toxicity. Calcineurin inhibitors (cyclosporine and tacrolimus) are effective in treating skin manifestations and, along with mycophenolate mofetil, are recommended for treating interstitial lung disease. Cyclophosphamide can be used in severe and rapidly progressive interstitial lung disease. Treatment with cyclophosphamide carries the risk of infertility. Intravenous immunoglobulins (IVIG) have also been shown to be beneficial in treating dermatomyositis.[5]
Biologics
Rituximab, an anti-CD 20 monoclonal antibody, is effective in treating myositis and myositis-related interstitial lung disease. Rituximab is preferred over cyclophosphamide by many clinicians due to its better tolerance and adverse effect profile. Although some patients might respond to anti-TNF agents, the studies on etanercept and infliximab have shown conflicting results. Few cases report tocilizumab (anti-IL-6 antibody), anakinra (anti-IL one antibody), alemtuzumab (anti-CD-52), tofacitinib, and ruxolitinib (JAK-inhibitors) as effective agents in treating autoimmune myositis. However, no confirmatory studies exist, and these drugs are used only in resistant cases.
Tailored physical exercise programs and rehabilitation under a physiatrist or physical therapist are shown to improve muscle strength and decrease disability, even in patients with acute inflammatory myopathy. Supportive treatment for PM/DM includes sunlight avoidance, the use of sunscreens in dermatomyositis, and aspiration precautions in patients with oropharyngeal myopathy.
Differential Diagnosis
The differential diagnosis of polymyositis and dermatomyositis (PM/DM) includes:
- Other idiopathic inflammatory myopathies (inclusion body myositis and immune-mediated necrotizing myopathy)
- Drug-induced myopathy
- Motor neuron disease
- Myasthenia gravis
- Lambert-Eaton syndrome
- Hypothyroidism
- Muscular dystrophy
- Myotonic dystrophy
- Amyotrophic lateral sclerosis
- Amyloid myopathy
- Sarcoid myopathy
- Diabetic amyotrophy
Enhancing Healthcare Team Outcomes
The management of polymyositis and dermatomyositis is with an interprofessional team because of the complexity of treatment. While the rheumatologist usually performs the initial treatment, the primary care provider and nurse practitioner often handle the patient follow-up. Corticosteroids are the initial therapy and cornerstone in managing both polymyositis and dermatomyositis (PM/DM).[4] There is no guideline available for the initial dosing or tapering of steroids, so the treatment should be under an experienced physician and based on clinical judgment. Patients and families should be educated about the disease and the treatment. Patients should be monitored for steroid-related adverse effects, particularly osteoporosis, steroid-induced myopathy, hypertension, hyperglycemia, and weight gain.
Steroid-sparing therapy like methotrexate and azathioprine (in patients with normal thiopurine methyltransferase (TPMT) activity) can be used in patients who develop adverse effects to steroids or those having trouble with tapering the steroids. They are also used in patients with inadequate responses to steroids. Side effects of these disease-modifying drugs need to be monitored.
Rituximab, anti-CD 20 monoclonal antibodies, is effective in treating myositis and myositis-related interstitial lung disease. Rituximab is preferred over cyclophosphamide by many clinicians due to its better tolerance and adverse effect profile. Pharmacists review medications for appropriateness, dosage, and interactions. They also provide patient education.
Finally, tailored physical exercise programs and rehabilitation under a physiatrist or physical therapist are shown to improve muscle strength and decrease disability, even in patients with acute inflammatory myopathy. Rehabilitation nurses monitor patient status and inform the team of major changes or issues.
Supportive treatment for PM/DM includes sunlight avoidance, the use of sunscreens in dermatomyositis, and aspiration precautions in patients with oro-pharyngeal myopathy. The outlook for patients with polymyositis and dermatomyositis is guarded.[6] [Level 5]