Continuing Education Activity
Ataxia-telangiectasia (A-T) is a rare neurodegenerative ataxia syndrome characterized by early childhood onset and autosomal recessive inheritance. The condition results from mutations in the ataxia telangiectasia mutated (ATM) gene located on chromosome 11q22-23 that cause neurological and systemic manifestations. Clinically, A-T is marked by cerebellar ataxia and the development of telangiectasias, dilated capillaries in the oculocutaneous region. As affected children begin to sit and walk, the onset of ataxia is evident through truncal instability.
The neurological symptoms associated with A-T include dysarthria, oculomotor apraxia, extrapyramidal symptoms, axonal neuropathy, and cognitive impairment. Beyond the neurological domain, individuals with A-T may experience immunological and endocrine abnormalities and an increased risk of neoplastic disease. Early childhood is a critical period for the emergence of neurological symptoms, with ataxia being the primary observable sign. The management of A-T necessitates an interprofessional team, and this activity aims to guide healthcare professionals in evaluating and improving care for patients with this complex condition.
Objectives:
Screen for the abnormal ataxia-telangiectasia mutated gene in the etiology of ataxia-telangiectasia.
Implement appropriate genetic testing to diagnose ataxia-telangiectasia.
Improve understanding of the pathophysiology of ataxia-telangiectasia.
Collaborate with all members of the interprofessional team, including age-appropriate primary care practitioners and specialists such as neurologists, endocrinologists, immunologists, and oncologists to provide efficient, comprehensive, and coordinated care.
Introduction
Ataxia-telangiectasia (A-T) is a rare inherited form of autosomal recessive neurodegenerative ataxia with onset in early childhood. Clinically, this syndrome manifests a combination of neurological and systemic symptoms due to the mutation of the ataxia-telangiectasia mutated (ATM) gene. Clinical hallmarks of A-T include cerebellar ataxia and dilated capillaries in the oculocutaneous region (telangiectasias).[1] Ataxia often presents in early childhood and is heralded by truncal instability as an affected child begins to sit and walk. A plethora of associated neurological and systemic signs and symptoms characterize A-T: neurologically, dysarthria, oculomotor apraxia, extrapyramidal symptoms, axonal neuropathy, and cognitive impairment are common; more broadly, individuals with A-T can experience immunological and endocrine abnormalities, and their risk of neoplastic disease is elevated. Serum levels of alpha-fetoprotein (AFP) are elevated.
Etiology
A-T is an autosomal recessive primary immunodeficiency (PID) disease caused by mutations in the ATM gene that encodes a serine/threonine protein kinase; the ATM gene is located on chromosome 11q22-23.[2] The mutated ATM gene causes aberrant repairing of the breaks in double-strand DNA. Because of this defect, cell response to various pathogenic triggers, such as ionizing radiation and alkylating agents, is impaired. Consequently, cell death occurs in susceptible tissues such as the cerebellum, and malignant proliferation arises.
In A-T, clinical manifestations include progressive cerebellar degeneration, oculocutaneous and other telangiectasias, variable immunodeficiency, radiation sensitivity, genome or chromosomal instability, cancer predisposition, chronic lung disease, segmental premature aging, and endocrine abnormalities. Recent studies now emphasize premature aging as an important driver of this disease. [3][4][5]
A-T is considered a congenital disorder with phenotypic heterogeneity, and the severity of symptoms corresponds to the severity of the underlying autosomal recessive mutations and the stage of disease.[3][6] Recent literature has also described A-T as one of four conditions demonstrating chromosomal instability syndromes, along with Bloom syndrome (BS), Fanconi anemia (FA), and Nijmegen breakage syndrome (NBS). These syndromes constitute a group of inherited disorders linked to chromosomal instability and breakage, either spontaneously or in response to DNA-damaging agents. The natural history of these syndromes is significant because each has been associated with various immunodeficiencies, infectious diseases, and the risk of developing specific types of malignancies. Early intervention to prevent complications relies on a deeper understanding of the roles of affected DNA repair pathways.[7][8]
Epidemiology
The global prevalence of A-T is estimated to be around 1 in 40,000 and 1 in 100,000 live births. [9] In some populations, the disease is as rare as 1:300,000.[10] In the United States, about 1% of the population is a carrier of a mutation in the ATM gene.[11] Males and females are equally affected by A-T. After Friedreich’s ataxia, A-T is the second most common childhood-onset neurodegenerative disorder of the cerebellum manifested as progressive ataxia and oculomotor apraxia and is often associated with extrapyramidal movement disorders.[12] However, A-T is the most common genetic ataxia with onset in the first decade. A significant founder effect has been reported among different populations, especially among the North African Jewish community.
Most of the ATM variants are found in the Caucasian population as well as Mexican pediatric patients. [13] Still, only a few studies focused on the genotype-phenotype correlation of A-T among Asians, especially in the Chinese population. However, a recent case report presented a Chinese patient with A-T who carries compound heterozygous variants in the ATM gene but implied that the diagnosis of A-T in China should be improved.[14]
Pathophysiology
Mutations of the ATM gene are responsible for A-T. Nonsense, frameshift, missense, and insertion-deletion mutations of the ATM gene have been described. In most cases, mutations lead to a truncated and, thus, nonfunctional protein product. Compound heterozygous mutations are not infrequent.
The mechanism underlying telangiectasia formation remains unknown. This condition results in dilated blood vessels and is one of the pathognomonic hallmarks of the disorder.
Irreversibleerebellar ataxia is commonly the first outcome in A-T due to selective cerebellar Purkinje neuronal degeneration. Recent reviews described how cerebellar Purkinje neurons become selectively vulnerable when all other cell types in the brain are affected by the same defects in ATM function.[15]
Another recent review also described the role of ATM in cerebellar pathology. The findings included:
- ATM functions in different cytoplasmic and mitochondrial processes critical in cellular homeostasis, aside from its established nuclear functions in DNA damage response circuits, with a focus on maintaining a homeostatic redox state.
- ATM functions in different types of neuronal and glial cells, such as cerebellar granule neurons, astrocytes, and microglial cells.[16]
ATM is involved in many different molecular mechanisms.[17] The gene’s protein product is essential for cellular DNA repair, cell cycle control, and cellular response to external triggers, such as oxidative damage, ionizing radiation, and alkylating agents. The protein itself is a serine/threonine kinase that affects a variety of downstream targets involved in important pathways for cellular protection against toxic insults. Loss of function of the ATM protein results in aberrant cellular proliferation due to unrepaired double-strand DNA breaks, leading to elevated cancer risk and radiosensitivity. Also, the impairment of cell cycle control can cause malformations, such as gonadal dysgenesis, found in patients with A-T. Moreover, ATM is necessary for immunoglobulin production and lymphoid cell survival. This explains why ATM mutations are responsible for a higher risk of tumors of the lymphatic system and autoimmune manifestations.
Recent studies have noted that A-T pathogenesis is not limited to the role of ATM in the DNA damage response pathway and that the gene has other functions, primarily in hematopoietic cells and neurons. Likewise, in cells ATM is not only involved with the nucleus to cope with DNA damage but is also associated with other organelles in the cytoplasm, suggesting that ATM may have a protective role. ATM adjusts the functions of organelles, such as mitochondria and peroxisomes, and ATM also regulates angiogenesis and glucose metabolisms.[3][18]
Recent studies have newly defined autophagy as a function of ATM. Findings showed that repression of ATM expression via siRNA inhibits autophagic flux at the autolysosome formation step and induces cell death in autophagy-inducing conditions. Therefore, ATM involvement in autolysosome formation and ATM inhibitors can be considered in cancer therapy.[19] The role of ATM in general autophagy, mitophagy, pexophagy, aggrephagy, and crosstalk between oxidative stress as an activator of ATM and its possible role as a primary regulator of these processes were also discussed in these studies.[20]
Another study identified that ATM orchestrated iron bioavailability through ferritinophagy and ferroptosis, the newly characterized form of programmed cell death driven by the lethal accumulation of lipid peroxides catalyzed by the intracellular bioactive iron. This condition holds great promise for therapeutic design against other therapy-resistant cancers.[21]
Histopathology
Autopsy cases of A-T mainly report degeneration of the cerebellar cortex. Purkinje cells and granular cells are especially affected. As A-T progresses, degeneration of other parts of the brain (specifically the brainstem) and the posterior and anterior horns of the spinal cord, develops.[22]
History and Physical
A-T is a complex disorder with clinical features that have variable severity throughout the natural history of the disease.
The classical form of A-T emerges early in the first decade of life. Neurological symptoms commonly appear in early childhood when children begin to sit or walk. Usually, ataxia is the first noticeable sign, heralded by truncal instability when toddlers are sitting or an unstable gait when walking. Patients with A-T often use wheelchairs by the age of 10.[9]
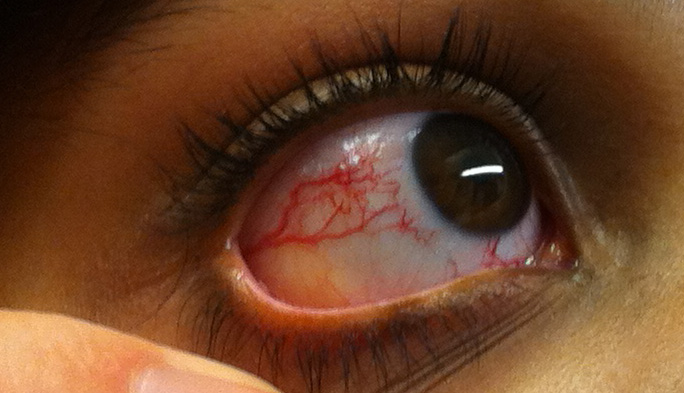
Telangiectasias are present in almost all cases. Ocular telangiectasias are pathognomonic for A-T and commonly appear after age 6, though they may be minimal or even absent altogether in milder variants. Ocular telangiectasias occur mainly in the conjunctiva and sun-exposed areas (such as the face and ears).[23] Telangiectasias can also be found in the brain and the bladder[24], but they generally do not progress, or are they prone to bleeding. Skin and visceral telangiectasias may develop with advancing age.[20] Premature aging, cutaneous atrophy, pigmentation changes (eg, café-au-lait macules and hypopigmented macules resembling vitiligo), melanocytic nevi, rashes, and hypertrichosis of the skin have also been reported.[25]
As A-T progresses, dysarthria and oculomotor apraxia may develop, affecting vision and reading skills and creating difficulty for affected children during their early school years. Nystagmus, saccadic intrusions, and hypometric saccades may also occur. Basal ganglia involvement is common, usually manifested by tremors, parkinsonism, chorea, dystonia, and myoclonus. Axonal polyneuropathy—usually subclinical—occurs as early as the first year of life.[26] A recent study reporting findings from a large national A-T cohort showed that neuropathy in A-T differs in the upper and lower extremities.[27]
Mild to moderate cognitive impairment has been reported and can affect different cognitive domains, such as language, memory, and executive function. In A-T, cognitive symptoms are variable, often with a wide spectrum in severity.[28] Recent studies have also reported consistent clinical manifestations of cerebellar cognitive affective syndrome (CCAS), such as impaired executive functioning, errors related to visual perceptual abilities, and emotional disorders such as anxiety and depression. The patients also presented with dyslalia and impairments in articulatory speech.[13][29]
Patients with classic A-T tend to be smaller and frequently experience height and weight faltering that begins in utero and continues relentlessly thereafter. Even if children appear to grow well at a young age, faltering begins early and becomes persistent.[30] In A-T, growth hormone deficiency is common, resulting in short stature, failure to grow, and gonadal failure, causing infertility.[31]
A-T patients have immunological abnormalities, including immunoglobulin, antibody deficiencies, and lymphopenia, commonly affecting about two-thirds of cases. Usually, patients present with low IgA and IgG levels, variable IgM, and low total CD4 cell count.[32][9] Moreover, individuals with A-T also have an increased predisposition for cancers, especially those of lymphoid origin. Pulmonary disease and problems with feeding, swallowing, and nutrition are typical, and there may also be dermatological and endocrine manifestations.[9] Frequent infections, especially severe sino-pulmonary infections, may indicate immunodeficiency in addition to autoimmune and chronic inflammatory diseases.[33] A-T patients are prone to symptomatic viral infections, especially in the setting of lower IgA and higher IgM titers, plus higher CD8+ counts.[34] Autoimmune deficiency generally does not progress.
About 25% to 30% of patients with A-T will develop neoplastic disease, most commonly leukemia and lymphoma, arising early in life. Later on, solid tumors such as breast and ovarian cancer, melanomas, and gastric and liver cancer can present.[35] Recent studies have identified A-T as a genetic risk factor for leukemia, and the tumor suppressor gene supported the presence of melanoma in-related cancer conditions.[36][37] Tumors can also be associated with severe radiosensitivity. In particular, x-ray and gamma-ray radiation, but not ultraviolet radiation, have been reported to be detrimental in patients with A-T and should be avoided.
Insulin resistance, frank diabetes, hypercholesterolemia, and steatosis can all occur, especially later in the course of the disease. Endocrine abnormalities are more frequent in females. Microcephaly has also been reported.
Variant Forms
Variant A-T is caused by mutations that allow some retained ATM kinase activity.[38] Some authors reported an adult-onset form of A-T, characterized by a much milder phenotype, that manifests later in life and progresses slowly. An intermediate phenotype that still presents during the first or second decade but has a more favorable course of progression has also been reported.[39] Milder phenotypes are frequently associated with dystonia.[39]
Ataxia-Telangiectasia-Like Phenotypes
Non-ATM genetic mutations have been found in patients with a phenotypic presentation similar to A-T, essentially amounting to formes frustes of A-T.[40]
Mutations in NBS1 or MRE11 have been associated with diseases that resemble A-T. Nijmegen breakage syndrome, which is caused by the mutation of the NBS1 gene, is characterized by an increased risk of cancer, microcephaly, and syndromic features of recurrent respiratory infections and intellectual disability. Ataxia and telangiectasia are not present.
The genetic mutations of the MRE11 gene cause ataxia-telangiectasia-like disorder 1 (ATLD1). ATLD1 can present with progressive ataxia, oculomotor apraxia, and dysarthria but without telangiectasias. Moreover, the age of onset is usually delayed compared to A-T, and the level of immunoglobulin in the peripheral blood is typically normal. An older study explored the eye movements characteristic of ATLD1, namely saccadic intrusions and cerebellar nystagmus along with fast and hypermetric saccades, linking them to damage of granule cell-parallel fiber-Purkinje cell synapses in the cerebellar vermis. The study explained this atypical phenotype based on a dual pathogenetic mechanism involving neurodevelopmental and neurodegenerative changes.[41]
Evaluation
The diagnosis of A-T can be challenging due to the rarity of the disorder. The diagnosis is usually based on the clinical presentation and findings from neuroimaging, laboratory tests, and genetics.
Clinical Picture
Early onset of ataxia in the first decade of life and oculomotor apraxia suggest the possibility of classical A-T and, if present, aid in focusing the diagnostic algorithm. The simultaneous presence of tumors may raise suspicion of A-T. The cohort of other symptoms, such as other neurological manifestations, and the involvement of the immunological, pulmonary, and endocrinological systems can help elucidate the diagnosis.
Telangiectasias
Telangiectasias are present in almost all A-T patients, representing a virtually pathognomonic sign to make the correct diagnosis. However, telangiectasias are not always easy to recognize. Moreover, they can present in unusual locations and go undetected if not attentively sought.
Neuroimaging
Cerebellar atrophy, mostly involving the vermis, is the cardinal radiological feature of A-T. Brain magnetic resonance imaging (MRI) is the best current imaging for these findings. Cerebellar atrophy becomes more prominent with age and may be absent early on. In older patients, white matter abnormalities have also been reported.
Ultrasound and Magnetic Resonance Imaging
Modern ultrasound and MRI are safe and efficient, and these modalities are now recommended as advantageous diagnostic imaging tools in monitoring children with syndromic inborn error of immunity and DNA instability syndromes, including A-T. The study detected the following abnormalities in the lungs, abdominal cavity, and lymph nodes:
- Hepatosplenomegaly
- Focal changes in the liver and spleen, with progressive mesenteric, visceral, and paraaortic lymphadenopathy corresponding to non-Hodgkin lymphoma
- Multiple subpleural pulmonary consolidations and B-line artifacts related to the interstitial-alveolar syndrome accompanied by pleural effusion, mediastinal masses, and bronchiectasis[42]
Laboratory Tests
Elevation of alpha-fetoprotein is a characteristic feature of A-T, even if it is not exclusive to this condition. Blood tests can also show a decrease in the total amount of IgG and IgA and variable levels of IgM, as well as decreased B-cell and T-cell numbers.[43]
Recently, a case described a patient with walking difficulties, frequent falls, and jerky head movements. Elevated serum alpha-fetoprotein levels suggested the possibility of A-T, which was confirmed by genetic testing of the ATM gene.[1]
Genetic Testing
The definitive diagnosis of A-T is reached by detecting homozygous or compound heterozygous mutation of the ATM gene. Genetic testing usually involves targeted gene sequencing or sequencing done as part of ataxia panels or whole-exome sequencing. In the case of a new gene variant, immunoblotting of the ATM protein can be performed to confirm whether the detected mutation causes a significant reduction of the levels of the ATM protein product.
Due to their kinase activity, the ATM protein can act as an amplifier, sensor, and signal transducer of DNA damage to the mediators of cell cycle arrest, apoptosis, and senescence. A recent study observed that classic A-T is related to homozygous ATM mutations, the complete absence of ATM kinase activity; truncation and nonsense mutations, loss of function mutation, non-conservative substitutions, frameshifts, and deletions are all possible. Meanwhile, in variant A-T, protein loss of function is incomplete, permitting some residual kinase activity. [44]
Treatment / Management
There is no cure for A-T as of the time of this review. The plethora of A-T clinical manifestations necessitates the involvement of an interprofessional team to address specific symptoms when they arise and to provide ongoing surveillance to minimize or prevent complications.[45]
Neurological Symptoms
Ataxia and other neurological manifestations can severely affect patients’ everyday life. Physical therapy and regular assessment for possible aids that may be needed are vital. One of the main goals of these assessments should be to prevent complications, such as falls. Children usually require support at school because of motor and cognitive impairment.
A recent case report was the first to support the benefits of physical therapy programs in A-T children. The study reported notable improvement in both body structure and function and activities and participation level, based on positive responses during motor performance, Gross Motor Function Measurement, Pediatric Berg Balance Scale, Trunk Control Measurement Scale, and participation as measured by the Life Habits Questionnaire and the Pediatric Quality of Life Inventory.[46]
Recent studies investigated acetyl-DL-leucine (ADLL), a modified form of an amino acid, as a possible treatment for A-T and reported improvement in ataxia, ocular stability, and quality of life (based on physical, emotional, social, and school functions) among AT patients.[47][48] Not all studies of ADLL have reported positive results, however. 34905009
A recent case report mentioned that an A-T patient received supportive therapy with amantadine and clonazepam, together with speech therapy. Still, the prognosis remained poor without targeted, disease-modifying treatment for A-T.[1]
Cancer and Radiosensitivity
Fastidious surveillance for breast and ovarian cancer can be lifesaving. Hematological tumors cannot be prevented, but regular monitoring facilitates early diagnosis. When tumors arise in A-T patients, their oncological care should follow standard, cancer-specific protocols, with one exception: ionizing radiation and some chemotherapeutic agents require careful monitoring due to an increase in A-T cell sensitivity.[47]
Immunological Disease
Treatment of recurrent infections and prophylactic antibiotic administration, when required, have been able to prolong life expectancy in patients with A-T. Intravenous immunoglobulin has been considered a good option for these patients. While inactivated vaccinations have not been associated with complications in patients with A-T, live vaccines may be contraindicated, especially with a low T-cell count.
Studies on nicotinamide riboside treatment with A-T showed improved ataxia scores and immunoglobulin levels. Antibiotic use and frequent hospitalizations due to infections were decreased by more than 90%. Immunological parameters in blood remained unchanged. No adverse effects occurred. Recently, a study hypothesized that early treatment would lead to even better outcomes, and given the absence of adverse effects, nicotinamide riboside use was encouraged for A-T patients.[51]
Complications secondary to restrictive lung disease must always be considered in case of anesthesia or surgery.[52] Pulmonary complications can be prevented with adequate surveillance of pulmonary function and prevention of recurrent respiratory infections.
Future Treatments
Antioxidants, antisense morpholino oligonucleotides, aminoglycoside antibiotics that can affect ATM protein function, and different small molecules targeting the ATM gene to address tumorigenesis have been tested. Most of these new therapeutic approaches are still under investigation.[53] Therapeutic trials are ongoing to test the efficacy of dexamethasone treatment through innovative delivery routes, such as patients’ autologous red blood cells loaded with dexamethasone (the ATTeST study). Recent studies also suggest that very low glucocorticoid doses may help improve A-T neurological symptomatology in some patients with A-T.[54] Moreover, short-term betamethasone treatment appears transiently to benefit patients with ataxia telangiectasia; however, long-term benefits and risks should be carefully considered since an adverse effect of transient adrenal dysfunction was observed in all cases.[55]
Differential Diagnosis
As noted previously, A-T has both variant forms and formes frustes, conditions that resemble A-T phenotypically but have different underlying molecular genetics.
The main categories of diseases that must be considered in the differential diagnosis of A-T are as follows:
Recessive Forms of Ataxia
Autosomal recessive disorders are more common in cases of consanguinity in the family. These disorders usually present early, and only the proband is affected. The list of autosomal recessive ataxia is extensive, and the incidence of these disorders varies in different populations. Among the autosomal recessive ataxias, Friedreich’s ataxia can be easily misdiagnosed as A-T: both conditions can present with early-onset cerebellar ataxia, polyneuropathy, and systemic involvement. The other autosomal recessive cerebellar ataxias can be subdivided into metabolic, congenital, degenerative, mitochondrial, and other conditions. The metabolic disorders include abetalipoproteinemia, cerebrotendinous xanthomatosis, and Refsum disease. The hereditary ataxias include Joubert syndrome, and the degenerative conditions include Charlevoix-Saguenay spastic ataxia and NARP syndrome (neuropathy, ataxia, retinitis pigmentosa), sensory ataxic neuropathy with dysarthria and ophthalmoplegia, infantile-onset spinocerebellar ataxia, and Marinesco-Sjogren syndrome.[49].
Ataxia with Oculomotor Apraxia
Oculomotor apraxia is a distinctive sign of A-T. However, there are other disorders where this can be found. Ataxia with oculomotor apraxia types 1 and 2 are autosomal recessive ataxias characterized by an early age of onset. The systemic symptoms commonly found in patients with A-T are not characteristically seen in these other forms.[50]
Prognosis
In A-T, overall survival can be improved by attentive care and careful ongoing surveillance screening to prevent recurrent infections and identify mass lesions as early as possible. Usually, patients affected by the classical form of A-T can reach early adulthood. The atypical forms of the disease typically present with a milder phenotype and a much longer life expectancy. Median survival was noted to be 19 to 25 years, with wide variability in 2 large cohorts.[51]
Complications
Many serious complications of A-T are possible, affecting both the quality and length of life.
A-T is a systemic and neurologic disease, potentially involving multiple organ systems. Complications are numerous and can include:
- Cognitive impairment
- Neurologic deficits
- Dysphagia
- Increased fall risk
- Diabetes
- Pulmonary disease
- Recurrent infections, including opportunistic infections
- Malignancy
Previous reviews have emphasized the prevention of secondary complications. For example, dysphagia can necessitate gastrostomy tube feedings to prevent pulmonary and nutritional complications of dysphagia. In situations requiring anesthesia, care must be taken to avoid impaired swallowing, aspiration, and infection. Surveillance for those with severe recurrent infections or undergoing immunomodulatory therapy includes monitoring pulmonary function and monitoring for early signs of malignancy, such as weight loss, bruising, localized pain, or swelling.[52]
The combination of recurrent pulmonary infections and restrictive pulmonary disease, which are common in these patients, can lead to bronchiectasis and interstitial lung diseases.[53]
Consultations
A recent study revealed that diabetes is a common finding among older A-T patients and often starts in puberty. The study recommended the need for an annual diabetes screening in patients older than 12 years.[54] Consultation with an endocrinologist or a diabetologist may be necessary. For surveillance reasons, collaboration with infectious disease, pulmonology, and oncology is recommended on a case-by-case basis. As noted previously, physical therapy may be beneficial.
Deterrence and Patient Education
Parents and patients with A-T should be educated regarding the importance of monitoring for malignancy and the prevention of recurrent infection. Medication adherence and the consequences of inadequate treatment should be explained in depth if antibiotics are prescribed. Due to the severe radiosensitivity of these patients, x-ray and gamma-ray exposure should be avoided.
Aside from typical health education and monitoring, genetic counseling is also recommended in previous reviews since A-T is inherited in an autosomal recessive manner. The family members should be informed that during conception, the siblings of an affected patient have a 25% chance of being affected, a 50% chance of being asymptomatic carriers, and a 25% chance of being unaffected and not carriers. ATM heterozygotes or carriers have a higher risk of developing cancer. Once the ATM variants have been determined in an affected family member, carrier testing for at-risk relatives, prenatal testing for a pregnancy at increased risk, and preimplantation genetic testing should be done.[52]
Pearls and Other Issues
Facts to keep in mind regarding A-T:
- Telangiectasias are small, widened blood vessels on the skin, which appear as small, reddish, or purple clusters.
- A-T is also known as cerebello-oculocutaneous telangiectasia or immunodeficiency with A-T. Eponymously, it is known as Louis Bar syndrome.
- Carriers of the ATM gene mutation have an increased risk of cancer and, as a result, reduced life expectancy. The incidence of radiosensitivity in carriers is still debated.
- Based on previous studies, all female carriers aged 40 to 50 and female ATM c.7271T>G mutation carriers aged 25 and older should be given intensified surveillance programs for breast cancer. Moreover, all carriers should be aware of lifestyle factors that may affect cardiovascular diseases and diabetes. [55]
Enhancing Healthcare Team Outcomes
A-T presents with a multiplicity of manifestations, meaning that the care of patients with A-T requires an interprofessional team that includes a geneticist, pediatrician or adult primary care practitioner, neurologist, oncologist, dermatologist, nurse practitioner, and infectious disease expert.[45]
Clinicians should be aware of the broad spectrum of clinical presentations of A-T.[56] No cure and no disease-modifying treatment is available for A-T. Therefore, addressing the specific symptoms associated with the disease and surveillance to prevent complications is crucial.
Since A-T is a rare, multisystem progressive condition usually observed in early childhood, A-T patients require coordinated multidisciplinary care to manage their complex needs and minimize the disease burden without a cure. There is a need for a team of guidance-specific nursing and allied healthcare providers within the community. A recent study mentioned therapeutic exercise, inspiratory muscle training, and early nutritional assessment and intervention are helpful.[27]
Only a few cases of variant AT with predominant movement disorder have been studied worldwide. Knowledge of these atypical presentations, such as genetically confirmed A-T with dystonia, helps in early diagnosis, thereby initiating management and counseling of the family as soon as possible.[57]