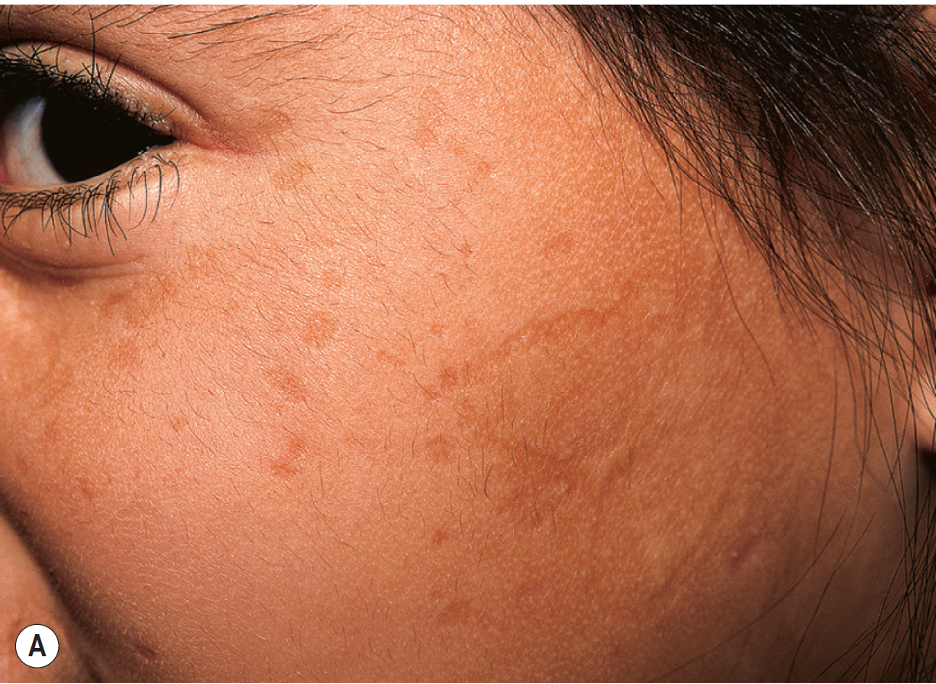
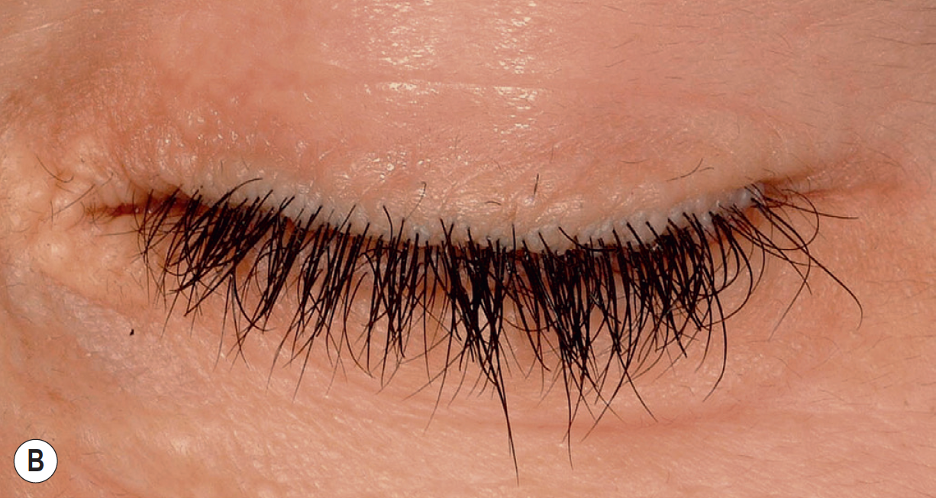
[1]
KATZENELLENBOGEN I,UNGAR H, Lipoid proteinosis; reinvestigation of a case previously reported by Urbach and Wiethe in 1929. Dermatologica. 1957 Jul;
[PubMed PMID: 13461538]
Level 3 (low-level) evidence
[2]
Teive HA,Ruschel E,Munhoz RP, Spontaneous intracerebral hemorrhage in Urbach-Wiethe disease. Neurology. 2013 Apr 30;
[PubMed PMID: 23628933]
[4]
Custódio Lima J,Nagasako CK,Montes CG,Barcelos IH,de Carvalho RB,Mesquita MA, Gastrointestinal involvement in lipoid proteinosis: a ten-year follow-up of a brazilian female patient. Case reports in medicine. 2014;
[PubMed PMID: 25045357]
Level 3 (low-level) evidence
[5]
Caccamo D,Jaen A,Telenta M,Varela E,Tiscornia O, Lipoid proteinosis of the small bowel. Archives of pathology
[PubMed PMID: 7514865]
[6]
Frenkel B,Vered M,Taicher S,Yarom N, Lipoid proteinosis unveiled by oral mucosal lesions: a comprehensive analysis of 137 cases. Clinical oral investigations. 2017 Sep;
[PubMed PMID: 27900487]
Level 3 (low-level) evidence
[7]
Nasir M,Latif A,Ajmal M,Qamar R,Naeem M,Hameed A, Molecular analysis of lipoid proteinosis: identification of a novel nonsense mutation in the ECM1 gene in a Pakistani family. Diagnostic pathology. 2011 Jul 26;
[PubMed PMID: 21791056]
[8]
Hamada T,Wessagowit V,South AP,Ashton GH,Chan I,Oyama N,Siriwattana A,Jewhasuchin P,Charuwichitratana S,Thappa DM,Jeevankumar B,Lenane P,Krafchik B,Kulthanan K,Shimizu H,Kaya TI,Erdal ME,Paradisi M,Paller AS,Seishima M,Hashimoto T,McGrath JA, Extracellular matrix protein 1 gene (ECM1) mutations in lipoid proteinosis and genotype-phenotype correlation. The Journal of investigative dermatology. 2003 Mar;
[PubMed PMID: 12603844]
[9]
Hamada T,McLean WH,Ramsay M,Ashton GH,Nanda A,Jenkins T,Edelstein I,South AP,Bleck O,Wessagowit V,Mallipeddi R,Orchard GE,Wan H,Dopping-Hepenstal PJ,Mellerio JE,Whittock NV,Munro CS,van Steensel MA,Steijlen PM,Ni J,Zhang L,Hashimoto T,Eady RA,McGrath JA, Lipoid proteinosis maps to 1q21 and is caused by mutations in the extracellular matrix protein 1 gene (ECM1). Human molecular genetics. 2002 Apr 1;
[PubMed PMID: 11929856]
[10]
Chan I,Liu L,Hamada T,Sethuraman G,McGrath JA, The molecular basis of lipoid proteinosis: mutations in extracellular matrix protein 1. Experimental dermatology. 2007 Nov;
[PubMed PMID: 17927570]
[11]
Di Giandomenico S,Masi R,Cassandrini D,El-Hachem M,De Vito R,Bruno C,Santorelli FM, Lipoid proteinosis: case report and review of the literature. Acta otorhinolaryngologica Italica : organo ufficiale della Societa italiana di otorinolaringologia e chirurgia cervico-facciale. 2006 Jun;
[PubMed PMID: 17063986]
Level 3 (low-level) evidence
[12]
Van Hougenhouck-Tulleken W,Chan I,Hamada T,Thornton H,Jenkins T,McLean WH,McGrath JA,Ramsay M, Clinical and molecular characterization of lipoid proteinosis in Namaqualand, South Africa. The British journal of dermatology. 2004 Aug;
[PubMed PMID: 15327549]
[13]
Nanda A,Alsaleh QA,Al-Sabah H,Ali AM,Anim JT, Lipoid proteinosis: report of four siblings and brief review of the literature. Pediatric dermatology. 2001 Jan-Feb;
[PubMed PMID: 11207965]
[14]
Youssefian L,Vahidnezhad H,Daneshpazhooh M,Abdollahzadeh S,Talari H,Khoshnevisan A,Chams-Davatchi C,Mobasher R,Li Q,Uitto J,Akhondzadeh S,Tabrizi M, Lipoid proteinosis: phenotypic heterogeneity in Iranian families with c.507delT mutation in ECM1. Experimental dermatology. 2015 Mar;
[PubMed PMID: 25529926]
[15]
Le Naour F,Hohenkirk L,Grolleau A,Misek DE,Lescure P,Geiger JD,Hanash S,Beretta L, Profiling changes in gene expression during differentiation and maturation of monocyte-derived dendritic cells using both oligonucleotide microarrays and proteomics. The Journal of biological chemistry. 2001 May 25;
[PubMed PMID: 11279020]
[16]
Han Z,Ni J,Smits P,Underhill CB,Xie B,Chen Y,Liu N,Tylzanowski P,Parmelee D,Feng P,Ding I,Gao F,Gentz R,Huylebroeck D,Merregaert J,Zhang L, Extracellular matrix protein 1 (ECM1) has angiogenic properties and is expressed by breast tumor cells. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2001 Apr;
[PubMed PMID: 11292659]
[17]
Rickman DS,Bobek MP,Misek DE,Kuick R,Blaivas M,Kurnit DM,Taylor J,Hanash SM, Distinctive molecular profiles of high-grade and low-grade gliomas based on oligonucleotide microarray analysis. Cancer research. 2001 Sep 15;
[PubMed PMID: 11559565]
[18]
Sercu S,Lambeir AM,Steenackers E,El Ghalbzouri A,Geentjens K,Sasaki T,Oyama N,Merregaert J, ECM1 interacts with fibulin-3 and the beta 3 chain of laminin 332 through its serum albumin subdomain-like 2 domain. Matrix biology : journal of the International Society for Matrix Biology. 2009 Apr;
[PubMed PMID: 19275936]
[19]
Fujimoto N,Terlizzi J,Aho S,Brittingham R,Fertala A,Oyama N,McGrath JA,Uitto J, Extracellular matrix protein 1 inhibits the activity of matrix metalloproteinase 9 through high-affinity protein/protein interactions. Experimental dermatology. 2006 Apr;
[PubMed PMID: 16512877]
[20]
Smits P,Poumay Y,Karperien M,Tylzanowski P,Wauters J,Huylebroeck D,Ponec M,Merregaert J, Differentiation-dependent alternative splicing and expression of the extracellular matrix protein 1 gene in human keratinocytes. The Journal of investigative dermatology. 2000 Apr;
[PubMed PMID: 10733679]
[21]
Sander CS,Sercu S,Ziemer M,Hipler UC,Elsner P,Thiele JJ,Merregaert J, Expression of extracellular matrix protein 1 (ECM1) in human skin is decreased by age and increased upon ultraviolet exposure. The British journal of dermatology. 2006 Feb;
[PubMed PMID: 16433788]
[23]
Olsen DR,Chu ML,Uitto J, Expression of basement membrane zone genes coding for type IV procollagen and laminin by human skin fibroblasts in vitro: elevated alpha 1 (IV) collagen mRNA levels in lipoid proteinosis. The Journal of investigative dermatology. 1988 May;
[PubMed PMID: 3361143]
[24]
Kowalewski C,Kozłowska A,Chan I,Górska M,Woźniak K,Jabłońska S,McGrath JA, Three-dimensional imaging reveals major changes in skin microvasculature in lipoid proteinosis and lichen sclerosus. Journal of dermatological science. 2005 Jun;
[PubMed PMID: 15927815]
[25]
Fabrizi G,Porfiri B,Borgioli M,Serri F, Urbach-Wiethe disease. Light and electron microscopic study. Journal of cutaneous pathology. 1980 Feb;
[PubMed PMID: 7358884]
[26]
Moy LS,Moy RL,Matsuoka LY,Ohta A,Uitto J, Lipoid proteinosis: ultrastructural and biochemical studies. Journal of the American Academy of Dermatology. 1987 Jun;
[PubMed PMID: 2439554]
[27]
Chan I,South AP,McGrath JA,Oyama N,Bhogal BS,Black MM,Hamada T, Rapid diagnosis of lipoid proteinosis using an anti-extracellular matrix protein 1 (ECM1) antibody. Journal of dermatological science. 2004 Aug;
[PubMed PMID: 15265527]
[28]
Sriwastava S,Desai A,Yuliati A,Watson CR,Sivaswamy L, A Child With a Hoarse Cry and Intracranial Calcification. Pediatric neurology. 2018 Oct;
[PubMed PMID: 30501889]
[30]
Savage MM,Crockett DM,McCabe BF, Lipoid proteinosis of the larynx: a cause of voice change in the infant and young child. International journal of pediatric otorhinolaryngology. 1988 Feb;
[PubMed PMID: 3372140]
[31]
Ally M,Kinshuck AJ,Sandison A,Sandhu GS, The management of laryngeal lipoid proteinosis. The Journal of laryngology and otology. 2018 Oct;
[PubMed PMID: 30099970]
[32]
Nayak S,Acharjya B, Lipoid proteinosis in a six-year-old child. Indian dermatology online journal. 2012 Jan;
[PubMed PMID: 23130256]
[33]
Belliveau MJ,Alkhotani A,Ali A, Moniliform Blepharosis of Lipoid Proteinosis. JAMA ophthalmology. 2015 Jul;
[PubMed PMID: 26158437]
[34]
Hamie L,Knio Z,Abbas O,Akel R,Bardawil T,Kibbi AG,Kurban M, Clinical clues early in the lives of individuals with lipoid proteinosis can determine the course of the disease. Clinical and experimental dermatology. 2017 Jun;
[PubMed PMID: 28244239]
[36]
Oguz Akarsu E,Dinçsoy Bir F,Baykal C,Taşdemir V,Kara B,Bebek N,Gürses C,Uyguner O,Baykan B, The Characteristics and Long-Term Course of Epilepsy in Lipoid Proteinosis: A Spectrum From Mild to Severe Seizures in Relation to ECM1 Mutations. Clinical EEG and neuroscience. 2018 May;
[PubMed PMID: 28434238]
[37]
Appenzeller S,Chaloult E,Velho P,de Souza EM,Araújo VZ,Cendes F,Li LM, Amygdalae calcifications associated with disease duration in lipoid proteinosis. Journal of neuroimaging : official journal of the American Society of Neuroimaging. 2006 Apr;
[PubMed PMID: 16629738]
[38]
Chandrasekaran S,Nanjundan M,Natarajan S,Ramadhas K, Radiologic presentation of lipoid proteinosis with symmetrical medial temporal lobe calcifications. Radiology case reports. 2015;
[PubMed PMID: 27398129]
Level 3 (low-level) evidence
[39]
Siebert M,Markowitsch HJ,Bartel P, Amygdala, affect and cognition: evidence from 10 patients with Urbach-Wiethe disease. Brain : a journal of neurology. 2003 Dec;
[PubMed PMID: 12937075]
[40]
Dertlioğlu SB,Çalık M,Çiçek D, Demographic, clinical, and radiologic signs and treatment responses of lipoid proteinosis patients: a 10-case series from Şanlıurfa. International journal of dermatology. 2014 Apr;
[PubMed PMID: 24320796]
Level 2 (mid-level) evidence
[41]
Fritsch PO, Retinoids in psoriasis and disorders of keratinization. Journal of the American Academy of Dermatology. 1992 Dec;
[PubMed PMID: 1460124]
[42]
Bakry OA,Samaka RM,Houla NS,Basha MA, Two Egyptian cases of lipoid proteinosis successfully treated with acitretin. Journal of dermatological case reports. 2014 Mar 31;
[PubMed PMID: 24748909]
Level 3 (low-level) evidence
[43]
Akoglu G,Karaduman A,Ergin S,Erkin G,Gokoz O,Unal OF,Hamada T, Clinical and histopathological response to acitretin therapy in lipoid proteinosis. The Journal of dermatological treatment. 2011 Jun;
[PubMed PMID: 20666665]
[44]
Toosi S,Ehsani AH, Treatment of lipoid proteinosis with acitretin: a case report. Journal of the European Academy of Dermatology and Venereology : JEADV. 2009 Apr;
[PubMed PMID: 18808438]
Level 3 (low-level) evidence
[45]
Gündüz O,Sahiner N,Atasoy P,Senyücel C, Acitretin treatment for lipoid proteinosis. Case reports in dermatological medicine. 2012;
[PubMed PMID: 23259080]
Level 3 (low-level) evidence
[46]
Wong CK,Lin CS, Remarkable response of lipoid proteinosis to oral dimethyl sulphoxide. The British journal of dermatology. 1988 Oct;
[PubMed PMID: 3191019]
[47]
Ozkaya-Bayazit E,Ozarmağan G,Baykal C,Uluğ T, [Oral DMSO therapy in 3 patients with lipoidproteinosis. Results of long-term therapy]. Der Hautarzt; Zeitschrift fur Dermatologie, Venerologie, und verwandte Gebiete. 1997 Jul;
[PubMed PMID: 9333627]
[48]
Kaya TI,Kokturk A,Tursen U,Ikizoglu G,Polat A, D-penicillamine treatment for lipoid proteinosis. Pediatric dermatology. 2002 Jul-Aug;
[PubMed PMID: 12220287]
[49]
Xu W,Wang L,Zhang L,Liu HG,Zan F,Hu R,Liu WB,Han DM, [Manifestation and treatment of lipoid proteinosis in larynx]. Zhonghua er bi yan hou tou jing wai ke za zhi = Chinese journal of otorhinolaryngology head and neck surgery. 2010 Apr;
[PubMed PMID: 20627049]
[50]
Harper JI,Duguid KP,Staughton RC,Moffat DA, Oropharyngeal and laryngeal lesions in lipoid proteinosis. The Journal of laryngology and otology. 1983 Sep;
[PubMed PMID: 6886551]
[51]
Rosenthal G,Lifshitz T,Monos T,Kachco L,Argov S, Carbon dioxide laser treatment for lipoid proteinosis (Urbach-Wiethe syndrome) involving the eyelids. The British journal of ophthalmology. 1997 Mar;
[PubMed PMID: 9135394]
[52]
Buchan NG,Kemble JV, Successful surgical treatment of lipoid proteinosis. The British journal of dermatology. 1974 May;
[PubMed PMID: 4833793]
[53]
Çalıskan E,Açıkgöz G,Tunca M,Koç E,Arca E,Akar A, Treatment of lipoid proteinosis with ablative Er:YAG laser resurfacing. Dermatologic therapy. 2015 Sep-Oct;
[PubMed PMID: 26031844]