Definition/Introduction
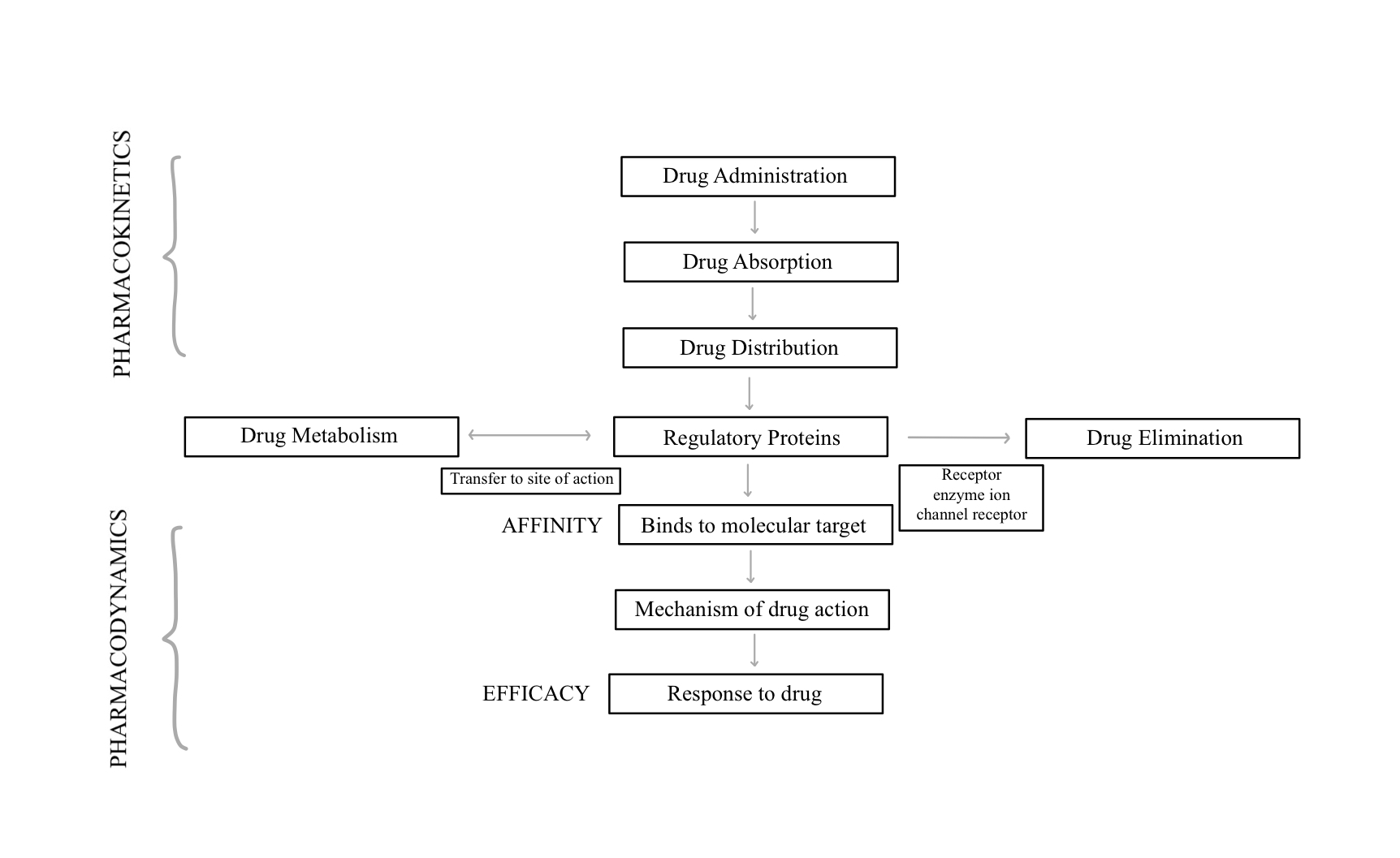
Pharmacokinetics is the study of a drug moiety or a compound as it moves through the body after its administration. It involves the processes of drug absorption, bioavailability, clearance, and distribution.[1] Although these processes are theoretically separate, from a practical standpoint in-vivo, they are all inter-connected. After the drug is absorbed from the site of administration, it is distributed to extracellular fluids.[2] High reserves of plasma protein-bound drugs can cause prolonged effects by creating a sustained release mechanism.[3]
Drug distribution is the disbursement of an unmetabolized drug as it moves through the body’s blood and tissues. The efficacy or toxicity of a drug depends on the distribution in specific tissues and in part explains the lack of correlation between plasma levels and the effects that are seen. Based on the molecular structure, drugs have variable distribution in different types of tissues such as fat, muscle, and brain. Unlike other tissues, the brain and testes are unique, as they contain membrane barriers making a drug significantly less susceptible to distribution.[4] Based on lipid and non-lipid solubility, drugs can be classified as lipophilic or hydrophilic described below.
Lipophiliic (Fat Soluble)
- Nonpolar compounds
- Easily diffuse across lipid bilayers of cell membranes
- Rx can be administered topically
- Free diffusion across the blood-brain barrier
- Biotransformed in the liver
- Excreted through the bile duct
Hydrophilic (Water Soluble)
- Polar compounds
- Cross lipid bilayers via facilitated transport (passive chemical diffusion across a cell membrane by ion channels or carriers)
- Eliminated by kidneys
Clinical Significance
As people age, the overall body water reduces. However, intracellular water remains relatively stable from the first month of life to adulthood. Higher doses of drugs per kilogram weight are required in younger children as they have a higher percentage of water.[5] Lipophilic drugs are more likely to distribute to areas of high lipid density.[6] Body fat varies with age, gender and genetics. Many drugs are bound to plasma proteins, and the most important drug-binding proteins include albumin and globulins. The concentration of these proteins varies with age, nutritional status, and disease.
Understanding drug distribution and pharmacokinetics (PK) is important for all clinicians prescribing medication, along with understanding the fundamentals of protein binding.[7] Only free and unbound drugs will pass from vascular spaces to tissues where a drug-receptor interaction will occur as well as the effect of the drug. Protein binding is not only affected by the concentration of protein but also the pH, metabolic abnormalities (hyperglycemia, uremia), and the presence of other chemicals that will compete for protein binding.
Competition for plasma binding can influence drug effects. For example, Aspirin and Warfarin are known to compete for the same plasma protein binding site. Administering both drugs at the same time will increase the unbound drug, thereby potentiating their effects and potentially lead to bleeding risk.[8] For a drug to be effectively eliminated by the kidney, the drug must be metabolized from a lipophilic molecule into a polar molecule. The liver produces a polar metabolite of the drug, using two unique sets of reactions known as phase I metabolism and phase II metabolism.[9]
Phase I metabolism involves what is known as the cytochrome P-450 system (CYP enzyme). CYP alters a drug in such a way so that it will be more amenable to combining with polar molecules. These reactions involve basic chemistry principles such as oxidation, reduction, or hydrolysis. Phase II metabolism is the process of adding a polar moiety to the drug, such as sulfate, acetate, or glucuronate. The addition of a polar moiety to a drug makes the drug water-soluble and available for excretion by the kidney.
Phases of Biotransformation
- Phase I reaction: The drug is first transformed into a polar metabolite via oxidation by the cytochrome P-450 system → allows phase II to occur.
- Phase II reactions: Involves the coupling of the metabolite with glucuronic acid, acetyl groups, sulfates, amino acids, or glutathione.
Types of Drug Kinetics
- Zero-order kinetics: The rate of metabolism/elimination remains constant and is independent of the concentration of a drug.
- First-order kinetics: The rate of metabolism/elimination is directly proportional to the plasma concentration of the drug.
Half-life (T1/2): The time required for a drug's plasma concentration to reach half of its initial value.
- After 4 half-lives > 90% of the drug is eliminated.
Drug clearance: The measure of the rate of drug elimination → the plasma volume that can be completely cleared of the drug in a given period of time.
Additionally, uremia not only affects protein binding, but kidneys also play a significant role in drug absorption, distribution, metabolism, and excretion (ADME). Renal dose adjustment is essential in moderate to severe renal failure. Important strategies for managing and drug dosing must be adjusted accordingly, and the risks must be weighed against the benefits.[10]
Several factors impact drug distribution. These factors include the concentration of drug transporters in blood, pH, perfusion, body water composition, body fat composition, and most certainly disease conditions (e.g., volume depletion, burns, third spacing). The majority of protein binding is relevant only when the drug is more than 90 percent protein bound. In the hypoalbuminemia state, which occurs in malnutrition and inflammation, there is a higher concentration of the unbound drugs. Body composition and metabolic factors also affect drug distribution. For example, during the last trimester of pregnancy, plasma volume expands, so there is an overall diluting effect on plasma proteins. There is also a change in adipose tissue. Additionally, pregnant women are frequently excluded in clinical trials related to drugs.[11] In a critically ill patient, drug distribution also changes due to deranged physiology, protein binding changes, fluid shifts, pH changes, and vascular organ perfusion.[12] Thus it could be useful to monitor drug levels in these conditions if possible.
The cytochrome P-450 system is a family of heme-containing enzymes found in the liver and intestinal tract. There are multiple forms of CYP enzymes. Some drugs can either induce or inhibit specific isoforms of the enzyme, affecting the ADME of a drug. A clinician must be aware of potential drug-drug interactions with CYP enzyme inducers and inhibitors and naturally occurring compounds that can alter the actions of CYP enzyme. Naturally occurring compounds include grapefruit juice, nicotine-containing products, and St. John’s wort.[13][14]
Below is a list of the major drugs that inhibit and induce the cytochrome P-450 system, as well as dugs that are a major substrate of the enzyme:
CYP1A2
- Inhibitors: amiodarone, cimetidine, ciprofloxacin, fluvoxamine
- Inducers: carbamazepine, phenobarbital, rifampin, tobacco
- Substrates: caffeine, clozapine, theophylline
CYP2C9
- Inhibitors: amiodarone, fluconazole, fluoxetine, metronidazole, ritonavir, trimethoprim/sulfamethoxazole
- Inducers: carbamazepine, phenobarbital, phenytoin, rifampin
- Substrates: carvedilol, celecoxib, glipizide, ibuprofen, irbesartan, losartan
CYP2C19
- Inhibitors: fluvoxamine, isoniazid, ritonavir
- Inducers: carbamazepine, phenytoin, rifampin
- Substrates: omeprazole, phenobarbital, phenytoin
CYP2D6
- Inhibitors: amiodarone, cimetidine, diphenhydramine, fluoxetine, paroxetine, quinidine, ritonavir, terbinafine
- Inducers: none
- Substrates: amitriptyline, carvedilol, codeine, donepezil, haloperidol, metoprolol, paroxetine, risperidone, tramadol
CYP2E1
- Inhibitors: none
- Inducers: ethanol, isoniazid, tobacco
- Substrates: acetaminophen, theophylline, verapamil
CYP3A4 and CYP3A5
- Inhibitors: clarithromycin, diltiazem, erythromycin, grapefruit juice, itraconazole, ketoconazole, nefazodone*, ritonavir, telithromycin, verapamil
- Inducers: carbamazepine, Hypericum perforatum, phenobarbital, phenytoin, rifampin
- Substrates: alprazolam, amlodipine, atorvastatin, cyclosporine, diazepam, estradiol, simvastatin, sildenafil, verapamil, zolpidem
Below is an additional list of common drug-drug interactions involving the cytochrome P-450 system that clinicians should be aware of:
Drug: amiodarone
CYP enzyme: CYP2C9 and CYP3A4 inhibitor
Drug-Drug Interaction: warfarin
Metabolizing Enzyme: CYP2C9
Side Effects: Increased risk of bleeding caused by increased warfarin level.
Drug: carbamazepine, phenobarbital, phenytoin
CYP enzyme: CYP3A4 inducer
Drug-Drug Interaction: Ethinyl estradiol-containing contraceptives
Metabolizing Enzyme: CYP3A4
Side Effects: Unplanned pregnancy caused by reduced estradiol level.
Drug: clarithromycin, erythromycin, telithromycin
CYP enzyme: CYP3A4 inhibitor
Drug-Drug Interaction: simvastatin, verapamil
Metabolizing Enzyme: CYP3A4
Side Effects: Myopathy or rhabdomyolysis caused by increased simvastatin level. Hypotension and QT interval prolongation caused by increased verapamil level.
Drug: diltiazem, verapamil
CYP enzyme: CYP3A4 inhibitor
Drug-Drug Interaction: prednisone
Metabolizing Enzyme: CYP3A4
Side Effects: Immunosuppression caused by increased prednisolone serum levels.
Drug: fluoxetine, paroxetine
CYP enzyme: CYP2D6 inhibitor
Drug-Drug Interaction: risperidone, tramadol
Metabolizing Enzyme: CYP2D6
Side Effects: Increased risk of extrapyramidal adverse effects caused by increased risperidone level.
Drug: Grapefruit juice
CYP enzyme: CYP3A4 inhibitor
Drug-Drug Interaction: buspirone
Metabolizing Enzyme: CYP3A4
Side Effects: Dizziness and serotonin syndrome caused by increased buspirone level.
Drug: metronidazole
CYP enzyme: CYP2C9 inhibitor
Drug-Drug Interaction: warfarin
Metabolizing Enzyme: CYP2C9
Side Effects: Increased risk of bleeding caused by increased warfarin level.
Drug: terbinafine
CYP enzyme: CYP2D6 inhibitor
Drug-Drug Interaction: amitriptyline
Metabolizing Enzyme: CYP2D6
Side Effects: Dry mouth, dizziness, and cardiac toxicity caused by a prolonged increase in amitriptyline and nortriptyline.
CYP = cytochrome P-450
Nursing, Allied Health, and Interprofessional Team Interventions
The interprofessional team and healthcare professionals, including laboratory technologists, pharmacists, nurses, and clinicians, need to all work together to ensure the safety and efficacy of administered drugs. After the clinician chooses the choice and dosage of a particular drug, the pharmacist should verify dosing, report any drug interactions, and take notice of special clinical situations that will influence drug levels and hence efficacy as well as adverse events (e.g., albumin levels, changes in weight, malnutrition, renal and hepatic function). In one study in chronic kidney disease patients, pharmacists identified 5302 drug-related problems and made 3160 recommendations with acceptance rates up to 95%.[15]
When possible and indicated plasma levels should be followed. Nurses play a critical role in drug administration and alerting the team regarding errors related to medication reconciliation.[16] This team collaboration is an essential part of patient safety in the inpatient and outpatient setting.