Continuing Education Activity
Keratoendotheliitis fugax hereditaria is a rare inflammatory genetic condition characterized by recurrent episodes of debilitating unilateral corneal and conjunctival hyperemia, corneal edema, visual impairment, corneal opacification, and photophobia that last for 2 to 5 days. Lacrimation, pain, colored halos, diplopia, a mild anterior chamber reaction, guttata-like changes (pseudoguttata), and a "gritty" feeling may also occur. The inheritance pattern of keratoendotheliitis fugax hereditariais is autosomal dominant. The pathology generally begins temporally before quickly progressing to surround the entirety of the cornea. In the acute phase, the pain or discomfort is often severe enough to disrupt sleep. This activity covers the genetics, pathophysiology, and evaluation of the patient with keratoendotheliitis fugax hereditaria. It also explores what is known about management, potential complications, and patient prognosis.
Objectives:
Recognize the most common aspects of patient history, as well as the unique clinical presentation and symptoms of keratoendotheliitis fugax hereditaria, distinguishing it from other ocular conditions with similar manifestations.
Differentiate between the acute inflammatory phase and the long-term corneal changes, understanding the temporal progression and triggers associated with each phase of the disease.
Implement timely and tailored treatment strategies during acute phases, utilizing anti-inflammatory agents, topical therapies, or novel interventions to mitigate symptoms and prevent long-term corneal damage.
Communicate observed physical exam findings accurately and effectively with patients, fellow healthcare professionals, and specialists, facilitating informed decision-making and collaborative care.
Introduction
Keratoendotheliitis fugax hereditaria is a rare inflammatory genetic condition characterized by recurrent episodes of debilitating unilateral corneal and conjunctival hyperemia, corneal edema, visual impairment, corneal opacification, and photophobia that last for 2 to 5 days.[1][2][3] Lacrimation, pain, colored halos, diplopia, a mild anterior chamber reaction, guttata-like changes (pseudoguttata), and a "gritty" feeling may also occur. The inheritance pattern of keratoendotheliitis fugax hereditariais is autosomal dominant. The pathology generally begins temporally before quickly progressing to surround the entirety of the cornea. In the acute phase, the pain or discomfort is often severe enough to disrupt sleep.
Although both the hyperemia and opacification generally resolve entirely within 2 to 4 days, localized opacity in the cornea may persist for weeks to months after the acute phase. Initial reports indicated that regardless of the duration of the corneal haze or the number of episodic incidents a person experiences, the opacification always completely remits, and vision returns to normal.[2] However, additional cases have shown individuals with permanent stromal opacification after numerous incidents.[1] These opacities, though evident on examination, may or may not affect visual acuity between episodes.
The disease was first described in 1964 and named in Valle's pedigree-based case report of Finnish individuals.[4] However, later reports hint that small differences in findings may indicate that the patients studied by Valle may have a closely related but slightly different pathology.
The frequency of these symptomatic inflammatory attacks varies by age demographic. Patients who are 15 to 20 years old experience the most frequent symptoms, occurring 1 to 8 times a year.[2][3] Attacks decrease in frequency and severity as the patient ages, though patients generally still experience moderate-to-severe events throughout their forties. However, by their mid-fifties, it appears patients are substantially less affected, with incidents causing comparatively mild symptoms and occurring less frequently.
Etiology
Keratoendotheliitis fugax hereditaria likely results from a missense mutation (c.61G>C) in the first exon of the nucleotide-binding domain, leucine-rich repeat family, pyrin domain-containing 3 gene (NLRP3). This gene codes for a protein called NLRP3 or cryopyrin. The mutation results in a partially positively charged histidine, replacing a negatively charged aspartic acid at the genetic locus.
As in many mutations, this change likely alters protein folding and function and may cause aberrant activation or dysregulation. This process may be problematic as NLRP3 combines with other protein molecules to assemble into inflammasomes, large complex structures that are involved and upregulated in the inflammation process.[3]
All 30 cases that have undergone genetic sequencing to assess for the mutation proved to be heterozygotes for the mutant gene. This finding may indicate that homozygosity may not be compatible with life or is too rare to have been observed and reported. Additionally, the severity and frequency of attacks have varied from generation to generation, indicating a potential for other genetic phenomena, such as incomplete penetrance, which has yet to be elucidated in this disease.
No risk factors are proven to evoke episodes of this sporadic disease, and seasonal changes have reportedly had no inciting effect on incident frequency. However, patients have anecdotally linked mild viral illnesses, cooler temperatures (ie, “sitting in the draft”), and the relief of physical or mental stress to the recurrence of symptomatic attacks.[1][2]
Additionally, several individuals in one pedigree were also affected by various collagen-based diseases. Since collagen and the neural crest cells from which the cornea derives are both mesodermal in origin, there may be an underlying association.
Epidemiology
No epidemiologic study has taken place to assess for cases of keratoendotheliitis fugax hereditaria. However, the associated NLRP3 gene mutation reportedly appears in about 0.02% of Finnish people and 0.01% of non-Finnish European individuals. Although all published case reports currently discuss families of Finnish ancestry, individuals of other hereditary lineages may carry the mutation.[3]
Pathophysiology
Several NLRP3 mutations have been grouped into a category of autosomal dominantly inherited autoinflammatory diseases termed cryopyrin-associated periodic syndrome (CAPS). This group of autoimmune inflammatory conditions also includes familial cold autoinflammatory syndrome (FCAS), chronic infantile neurological cutaneous and articular (CINCA) syndrome, Muckle-Wells syndrome (MWS), and neonatal-onset multisystem inflammatory disease (NOMID). However, these mutations are at different genetic loci.[3][5]
These syndromes have ocular symptoms that overlap with keratoendotheliitis fugax hereditaria, including keratitis, conjunctivitis, episcleritis, and clouding of corneal stroma; corneal involvement appears in 40% of patients with CAPS syndromes. However, unique characteristics of these diseases separate them from keratoendotheliitis fugax hereditaria, including papilledema, uveitis, and corneal neovascularization, in addition to their systemic symptoms such as fever.[5][6][7][8]
The NLR family of proteins encodes for inflammatory-based molecules in numerous cell lines. NLRP3 is expressed specifically in peripheral leukocytes as well as ocular structures such as lens epithelium and corneal endothelium and plays a central role in the activation of the NLRP3 inflammasome.[3][9][10][11] The inflammasome is a proinflammatory complex of proteins assembled, that regulates proinflammatory processes and caspase (proapoptotic) activity. Understandably, such processes are under tight regulation on a cellular level. Activation of this inflammasome is associated with several autoimmune conditions, including multiple sclerosis, systemic lupus erythematosus, rheumatoid arthritis, and irritable bowel disease.[11][12]
It has been shown that activation of the NLRP3 inflammasome is associated with corneal inflammation, and inhibition of NLRP3 has been shown to improve corneal wound healing.[9][13][14] Additionally, mice with inactivated NLRP3 genes were shown to have decreased corneal scarring and significantly decreased opacity grade after sterile burn injury compared with mice with functional NLRP3 genes.[15] Accordingly, aberrant activation of the NLRP3 inflammasome due to a mutation may cause inflammatory signs and symptoms with resultant corneal opacification or scarring. The c.61G>C NLRP3 mutation that causes keratoendotheliitis fugax hereditaria may lead to defects in the structure of the protein. In turn, the poorly functioning protein may allow for premature or easy activation of the inflammasome and may therefore be responsible for the sporadic inflammatory response that typifies the disease.
As a result of the inflammatory effects, swelling and edema may develop in the corneal stroma. Proposals exist, suggesting that this occurs secondarily to a vasogenic process, thus resulting in visual impairment and symptomatic halos and diplopia.[2] This intracellular and intercellular edema and subsequent "looseness" have also been proposed to cause the pseudoguttata observed in imaging, and are less prominent outside of acute episodes.[1][16][17][18] Pseudoguttata appear similar to guttata but have distinct histopathological differences and resolve, unlike the permanent Descemet membrane effects seen in true guttata. In addition to pseudoguttata, there is also a proposal that a decreased number of endothelial cells seen in some individuals is the result of cell death secondary to the observed inflammation.[1] However, not all reports have shown this observed decrease in cell count.[3]
History and Physical
The presentation of endotheliitis fugax hereditaria can vary, though characteristically, patients will complain of an incapacitating painful red eye with decreased visual acuity, which may begin with a foreign body sensation or stiff neck before quickly progressing. There may be associated lacrimation and congestion of the ipsilateral nostril with a watery discharge that occurs in about 50% of patients. The symptoms are typically unilateral, but bilateral or sequentially unilateral (ie, one eye develops symptoms immediately after the initial flare resolves in the opposing eye) cases are not uncommon.[2] As the initial pain and hyperemia resolve, the patient's remaining complaint will be a decreased visual acuity from resultant corneal edema and haze.[1]
The disease typically presents around age 10 to 11 (although there are reports that it develops as early as 3 years of age and as late as 28), and the possibility of an initial attack merits strong consideration in younger children with a positive family history. In older individuals, a history of similar events that occur between 1 and 6 times a year may be present, though reports exist of spontaneous mutations. As the disease is genetically linked, family history is essential to making a correct diagnosis. A complete hereditary history will likely show the disease inherited in an autosomal dominant fashion, although spontaneous mutations can occur.[2][3]
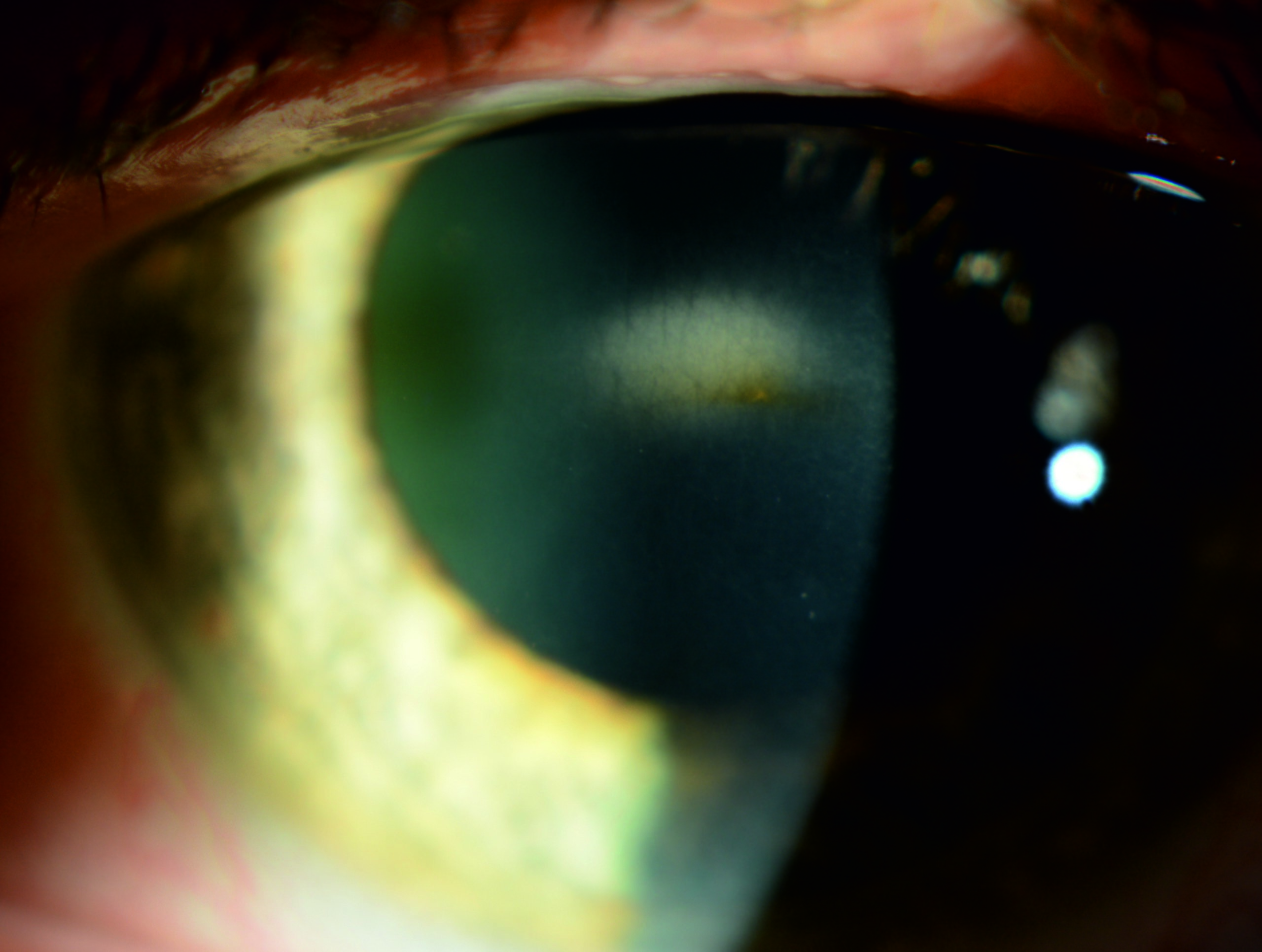
On slit-lamp examination in the acute phase, conjunctival injection and corneal edema will likely be present. An opacity in the central stroma, with or without keratic precipitates on the posterior surface of the cornea, is a common aspect of the disease and may be seen.[1][2] Furthermore, the endothelium's "beaten silver" appearance may be present, and central pseudoguttata (edematous endothelial cells) have been described. Corneal thickness may increase by 5% to 14% during an acute attack, though one patient presented with thinner areas of the cornea in the center.
However, if the patient presents following the initial acute phase (ie, 2 to 4 days following the onset of symptoms), the only visible sequela of the disease still likely to be present is the stromal opacification or posterior precipitates.[1][2] This opacity may be a permanent aspect of the disease and can be vision-effecting outside of acute incidents. Other findings include occasional corneal erosions; one patient had prominent iris atrophy.[3] Tomography, ocular coherence tomography (OCT), and corneal thickness are typically unremarkable between attacks, except for patients with stromal scarring and opacification.
Endothelial specular photography may reveal a decreased number of endothelial cells.[1][3] During an attack, specular imaging may show normal hexagonal cells with adjacent large, black, nonreflecting areas between them (pseudoguttata), and the changes may be apparent. Between incidents, marked pleomorphism and black spots in the center of cells may be visible in an otherwise normal-appearing endothelial mosaic.
Evaluation
Although genetic testing is confirmatory, such methods may not be practical or necessary for all patients, especially those with known family history and characteristic symptoms. However, spontaneous cases in which the diagnosis is more ambiguous may benefit from genetic confirmation.
Evaluation of the eye differs on whether the patient is experiencing an acute attack. This phenomenon may be difficult to assess in the clinic due to the short duration of symptoms. In an acute attack, there may be an increase in the corneal thickness (which may be associated with pseudoguttata formation).[2][19] Pseudoguttata may present using confocal, specular, or light microscopy.[17]
Imaging and testing can rule out other potential causes outside of an attack. Intraocular pressure is expected to be stable, even in acute disease. Aqueous humor will be clear since the pathology is primarily in the cornea. Additionally, synechiae do not appear to develop.[2] As noted previously, OCT is not particularly helpful, and tomography and topography are typically normal outside of an acute episode. Since specular photographic changes are evident even in between attacks, this imaging modality may be the most useful in determining patients who may have the disease when it is not an acute episode.[1]
Treatment / Management
As the underlying etiology of keratoendotheliitis fugax hereditaria is genetic, and the pathophysiology is incompletely understood, treatment is mainly supportive based on published case reports. As reported, the best treatment is topical corticosteroids, though the frequency and dose have not been published. However, not all patients respond to topical steroid treatment. Also, reports exist that oral antihistamines have been of symptomatic benefit to patients, and drowsiness typically associated with some drugs in this class may help patients who struggle to sleep through their symptoms.[2] Topical or oral nonsteroidal antiinflammatory drugs (NSAIDs) are additional modalities that can relieve the pain associated with attacks.[1][3]
Regardless of the selected treatment choice, therapy should begin immediately as early intervention limits symptoms. Delaying treatment until corneal inflammation is apparent seems not to affect recovery. Many patients have reported that early treatment with topical steroid drops or ointments may stop symptoms from progressing or alleviate symptoms more rapidly.[1][2][3] No invasive or surgical management has been reported.
Regarding visual acuity, some patients have found more satisfactory results with rigid contact lenses than with glasses.[3]
As NLRP3 activation plays a central role in inflammation, the mutation that causes keratoendotheliitis fugax hereditaria may cause dysregulation of the protein and resultant inflammation, modulators that inhibit activating upstream effects or subsequent proinflammatory downstream effects may be of benefit in the treatment of this disease. Inhibitors of NLRP3 effects, including MCC950, beta-hydroxybutyrate, type 1 interferon, and interferon beta, resveratrol, arglabin, CB2R agonists, and MicroRNA-223 may, therefore, prove beneficial in the treatment or prevention of acute attacks and prevent corneal scarring.[9][11][12] Many of these agents have already demonstrated a capacity to limit the inflammatory response in various inflammatory ocular pathologies and cell lines with varying effects.[20][21][22][23] More research is needed to further understand the effect of these inhibitors on corneal cells and keratoendotheliitis fugax hereditaria specifically.
Differential Diagnosis
Cases with an established genetic lineage and typical symptoms may be diagnosed clinically, but sporadic cases are especially challenging to diagnose. As keratoendotheliitis fugax hereditaria in the acute phase dies down, clinicians may incorrectly diagnose the residual inflammation as acute anterior uveitis.[3] Differential diagnoses, therefore, generally revolve around other inflammatory eye diseases, including uveitis or endotheliitis, with or without pseudoguttata. The etiologies may include infectious (eg, bacterial, viral, fungal, etc), autoimmune, drug-induced, or other rare genetic causes.[17][24]
- Iridocorneal endothelial syndrome
- Anterior uveitis
- Chandler syndrome
- Angle-closure glaucoma
- Relative anterior microphthalmos
- Brown-McLean syndrome,
- Posterior polymorphous corneal dystrophy
Prognosis
The prognosis for patients with keratoendotheliitis fugax hereditaria is fair. Although the frequency and severity of the condition lessen with time, stromal scarring, seen in 50% of patients, may permanently affect vision. These permanent opacifications seem to increase with repeated attacks as they are denser and more debilitating in older patients.[3]
It is possible that other individuals with less severe forms of the mutation exist or that less severe expressivity in an individual may result in mild or subclinical symptoms. If these cases exist, the prognosis is likely good to excellent and may not need treatment.
Complications
Aside from the pain and discomfort that may be associated with acute episodes, the primary long-term complication is a decline in visual acuity. An increased number of attacks may contribute to permanent corneal scarring and a decrease in the quality of life.[3]
Deterrence and Patient Education
As the disease is genetic, and without an identified trigger for incidental episodes, there are no known preventative or prophylactic steps that patients can take to limit or decrease the frequency, duration, or severity of attacks. Patients should be educated that any episodes of decreased vision should warrant an ophthalmologic examination.
Pearls and Other Issues
- Keratoendotheliitis fugax hereditaria is an autosomal dominantly inherited condition that presents with sporadic episodes of pain, conjunctival hyperemia, and corneal edema, which can lead to decreased visual acuity. These incidents last 2 to 5 days.
- Common findings include corneal opacity, keratic precipitates, pseudoguttata, and a "beaten silver" appearance of the endothelium.
- Though sporadic cases exist, patients are generally children to teenagers of Finnish descent with a strong family history.
- Episodes are self-resolving, with minimal acute complications; however, permanent corneal scarring and decreased visual acuity may develop after recurrences.
- The condition is usually unilateral, though bilateral episodes have also been reported.
- The condition is likely the result of an NLRP3 gene mutation that causes easily activated inflammatory responses.
- Treatment is aimed at symptomatic relief using a topical steroid, topical NSAIDs, or oral antihistamine medications.
- Molecules that inhibit the effects of NLRP3 may help to treat this disease, though more research is necessary to understand the effect of these drugs on corneal cells.
Enhancing Healthcare Team Outcomes
Patients with suspected keratoendotheliitis fugax hereditaria require a referral to an ophthalmologist familiar with the disease. Genetic counselors or geneticists should also be included in the healthcare team to provide information and resources to these patients. Prescriptions of topical or oral medications may be of some benefit to these individuals. As newer immunologic inflammatory inhibitors become available as pharmaceutical options, the healthcare team should also include a pharmacist familiar with the pharmacokinetics of these drugs to help optimize outcomes.