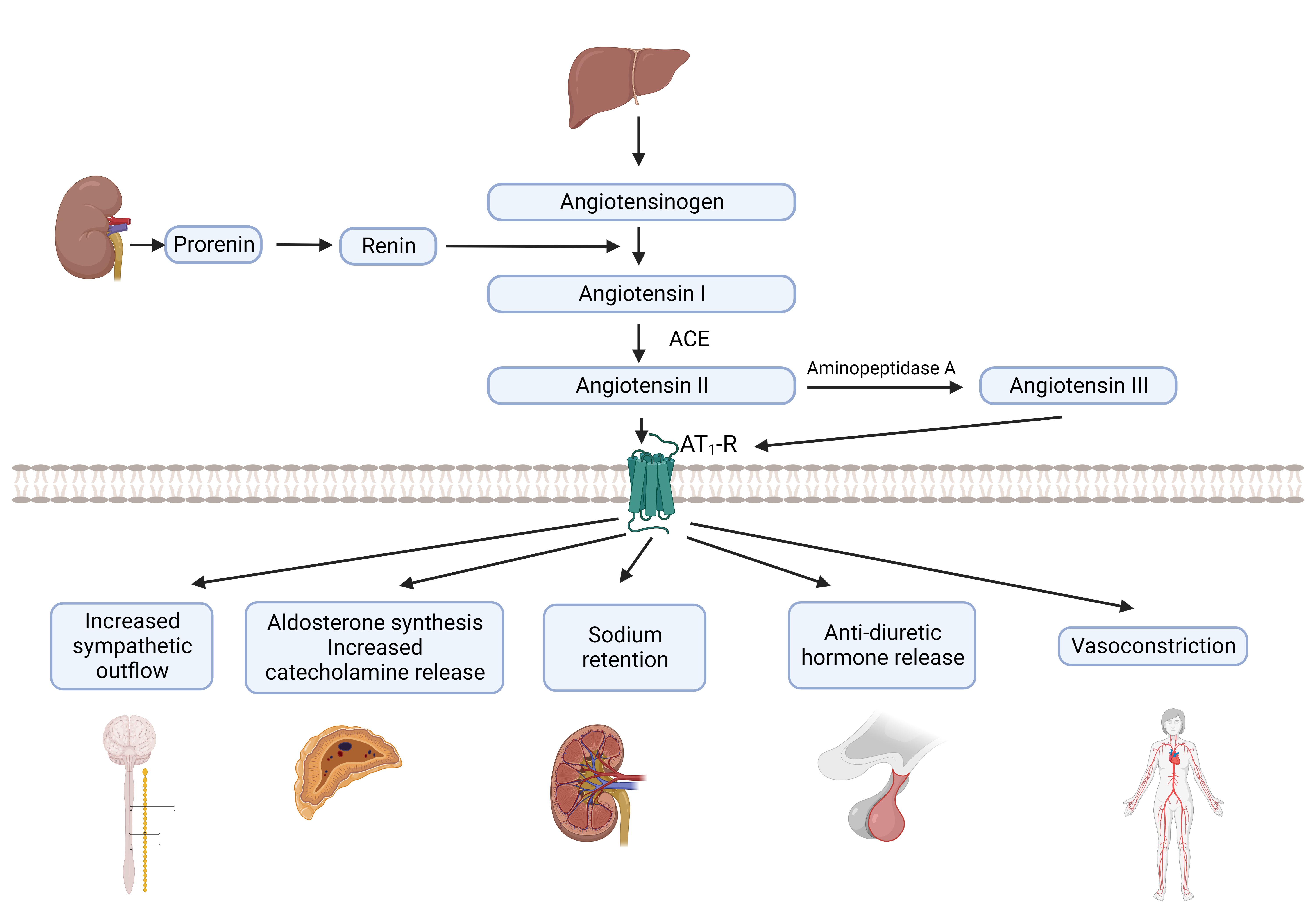
[1]
Almutlaq M, Alamro AA, Alroqi F, Barhoumi T. Classical and Counter-Regulatory Renin-Angiotensin System: Potential Key Roles in COVID-19 Pathophysiology. CJC open. 2021 Aug:3(8):1060-1074. doi: 10.1016/j.cjco.2021.04.004. Epub 2021 Apr 15
[PubMed PMID: 33875979]
Level 2 (mid-level) evidence
[2]
Wu CH, Mohammadmoradi S, Chen JZ, Sawada H, Daugherty A, Lu HS. Renin-Angiotensin System and Cardiovascular Functions. Arteriosclerosis, thrombosis, and vascular biology. 2018 Jul:38(7):e108-e116. doi: 10.1161/ATVBAHA.118.311282. Epub
[PubMed PMID: 29950386]
[3]
Santos RAS, Oudit GY, Verano-Braga T, Canta G, Steckelings UM, Bader M. The renin-angiotensin system: going beyond the classical paradigms. American journal of physiology. Heart and circulatory physiology. 2019 May 1:316(5):H958-H970. doi: 10.1152/ajpheart.00723.2018. Epub 2019 Feb 1
[PubMed PMID: 30707614]
[4]
Engeli S, Negrel R, Sharma AM. Physiology and pathophysiology of the adipose tissue renin-angiotensin system. Hypertension (Dallas, Tex. : 1979). 2000 Jun:35(6):1270-7
[PubMed PMID: 10856276]
[5]
Atlas SA. The renin-angiotensin aldosterone system: pathophysiological role and pharmacologic inhibition. Journal of managed care pharmacy : JMCP. 2007 Oct:13(8 Suppl B):9-20
[PubMed PMID: 17970613]
[6]
Reudelhuber TL, Ramla D, Chiu L, Mercure C, Seidah NG. Proteolytic processing of human prorenin in renal and non-renal tissues. Kidney international. 1994 Dec:46(6):1522-4
[PubMed PMID: 7699995]
[7]
Neves FA, Duncan KG, Baxter JD. Cathepsin B is a prorenin processing enzyme. Hypertension (Dallas, Tex. : 1979). 1996 Mar:27(3 Pt 2):514-7
[PubMed PMID: 8613195]
[8]
Schweda F, Friis U, Wagner C, Skott O, Kurtz A. Renin release. Physiology (Bethesda, Md.). 2007 Oct:22():310-9
[PubMed PMID: 17928544]
[9]
Kurtz A. Control of renin synthesis and secretion. American journal of hypertension. 2012 Aug:25(8):839-47. doi: 10.1038/ajh.2011.246. Epub 2012 Jan 12
[PubMed PMID: 22237158]
[10]
Kurtz A. Renin release: sites, mechanisms, and control. Annual review of physiology. 2011:73():377-99. doi: 10.1146/annurev-physiol-012110-142238. Epub
[PubMed PMID: 20936939]
[11]
Skrabal F. Half-life of plasma renin activity in normal subjects and in malignant hypertension. Klinische Wochenschrift. 1974 Dec 15:52(24):1173-4
[PubMed PMID: 4456013]
[12]
SKEGGS LT Jr, KAHN JR, LENTZ K, SHUMWAY NP. The preparation, purification, and amino acid sequence of a polypeptide renin substrate. The Journal of experimental medicine. 1957 Sep 1:106(3):439-53
[PubMed PMID: 13463253]
[13]
Laghlam D, Jozwiak M, Nguyen LS. Renin-Angiotensin-Aldosterone System and Immunomodulation: A State-of-the-Art Review. Cells. 2021 Jul 13:10(7):. doi: 10.3390/cells10071767. Epub 2021 Jul 13
[PubMed PMID: 34359936]
[14]
Studdy PR, Lapworth R, Bird R. Angiotensin-converting enzyme and its clinical significance--a review. Journal of clinical pathology. 1983 Aug:36(8):938-47
[PubMed PMID: 6308066]
[15]
Guo DF, Sun YL, Hamet P, Inagami T. The angiotensin II type 1 receptor and receptor-associated proteins. Cell research. 2001 Sep:11(3):165-80
[PubMed PMID: 11642401]
[16]
van Kats JP, de Lannoy LM, Jan Danser AH, van Meegen JR, Verdouw PD, Schalekamp MA. Angiotensin II type 1 (AT1) receptor-mediated accumulation of angiotensin II in tissues and its intracellular half-life in vivo. Hypertension (Dallas, Tex. : 1979). 1997 Jul:30(1 Pt 1):42-9
[PubMed PMID: 9231819]
[17]
Yatabe J, Yoneda M, Yatabe MS, Watanabe T, Felder RA, Jose PA, Sanada H. Angiotensin III stimulates aldosterone secretion from adrenal gland partially via angiotensin II type 2 receptor but not angiotensin II type 1 receptor. Endocrinology. 2011 Apr:152(4):1582-8. doi: 10.1210/en.2010-1070. Epub 2011 Feb 8
[PubMed PMID: 21303953]
[18]
Harrison-Bernard LM. The renal renin-angiotensin system. Advances in physiology education. 2009 Dec:33(4):270-4. doi: 10.1152/advan.00049.2009. Epub
[PubMed PMID: 19948673]
Level 3 (low-level) evidence
[19]
Gupta P, Franco-Saenz R, Mulrow PJ. Locally generated angiotensin II in the adrenal gland regulates basal, corticotropin-, and potassium-stimulated aldosterone secretion. Hypertension (Dallas, Tex. : 1979). 1995 Mar:25(3):443-8
[PubMed PMID: 7875770]
[20]
Nogueira EF, Xing Y, Morris CA, Rainey WE. Role of angiotensin II-induced rapid response genes in the regulation of enzymes needed for aldosterone synthesis. Journal of molecular endocrinology. 2009 Apr:42(4):319-30. doi: 10.1677/JME-08-0112. Epub 2009 Jan 21
[PubMed PMID: 19158234]
[21]
Cano A, Miller RT, Alpern RJ, Preisig PA. Angiotensin II stimulation of Na-H antiporter activity is cAMP independent in OKP cells. The American journal of physiology. 1994 Jun:266(6 Pt 1):C1603-8
[PubMed PMID: 8023891]
[22]
Reid IA. Interactions between ANG II, sympathetic nervous system, and baroreceptor reflexes in regulation of blood pressure. The American journal of physiology. 1992 Jun:262(6 Pt 1):E763-78
[PubMed PMID: 1616014]
[23]
Qadri F, Culman J, Veltmar A, Maas K, Rascher W, Unger T. Angiotensin II-induced vasopressin release is mediated through alpha-1 adrenoceptors and angiotensin II AT1 receptors in the supraoptic nucleus. The Journal of pharmacology and experimental therapeutics. 1993 Nov:267(2):567-74
[PubMed PMID: 8246129]
[24]
Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. American journal of physiology. Cell physiology. 2007 Jan:292(1):C82-97
[PubMed PMID: 16870827]
[25]
Rajagopalan S, Kurz S, Münzel T, Tarpey M, Freeman BA, Griendling KK, Harrison DG. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. The Journal of clinical investigation. 1996 Apr 15:97(8):1916-23
[PubMed PMID: 8621776]
[26]
Dzau VJ. Theodore Cooper Lecture: Tissue angiotensin and pathobiology of vascular disease: a unifying hypothesis. Hypertension (Dallas, Tex. : 1979). 2001 Apr:37(4):1047-52
[PubMed PMID: 11304501]
[27]
Schieffer B, Schieffer E, Hilfiker-Kleiner D, Hilfiker A, Kovanen PT, Kaartinen M, Nussberger J, Harringer W, Drexler H. Expression of angiotensin II and interleukin 6 in human coronary atherosclerotic plaques: potential implications for inflammation and plaque instability. Circulation. 2000 Mar 28:101(12):1372-8
[PubMed PMID: 10736279]
[28]
AbdAlla S, Lother H, Abdel-tawab AM, Quitterer U. The angiotensin II AT2 receptor is an AT1 receptor antagonist. The Journal of biological chemistry. 2001 Oct 26:276(43):39721-6
[PubMed PMID: 11507095]
[29]
Ferrario CM. Role of angiotensin II in cardiovascular disease therapeutic implications of more than a century of research. Journal of the renin-angiotensin-aldosterone system : JRAAS. 2006 Mar:7(1):3-14
[PubMed PMID: 17083068]
[30]
Xu Z, Li W, Han J, Zou C, Huang W, Yu W, Shan X, Lum H, Li X, Liang G. Angiotensin II induces kidney inflammatory injury and fibrosis through binding to myeloid differentiation protein-2 (MD2). Scientific reports. 2017 Mar 21:7():44911. doi: 10.1038/srep44911. Epub 2017 Mar 21
[PubMed PMID: 28322341]
[31]
Karnik SS, Unal H, Kemp JR, Tirupula KC, Eguchi S, Vanderheyden PM, Thomas WG. International Union of Basic and Clinical Pharmacology. XCIX. Angiotensin Receptors: Interpreters of Pathophysiological Angiotensinergic Stimuli [corrected]. Pharmacological reviews. 2015 Oct:67(4):754-819. doi: 10.1124/pr.114.010454. Epub
[PubMed PMID: 26315714]
[32]
Carey RM, Wang ZQ, Siragy HM. Role of the angiotensin type 2 receptor in the regulation of blood pressure and renal function. Hypertension (Dallas, Tex. : 1979). 2000 Jan:35(1 Pt 2):155-63
[PubMed PMID: 10642292]
[33]
Zhang H, Han GW, Batyuk A, Ishchenko A, White KL, Patel N, Sadybekov A, Zamlynny B, Rudd MT, Hollenstein K, Tolstikova A, White TA, Hunter MS, Weierstall U, Liu W, Babaoglu K, Moore EL, Katz RD, Shipman JM, Garcia-Calvo M, Sharma S, Sheth P, Soisson SM, Stevens RC, Katritch V, Cherezov V. Structural basis for selectivity and diversity in angiotensin II receptors. Nature. 2017 Apr 20:544(7650):327-332. doi: 10.1038/nature22035. Epub 2017 Apr 5
[PubMed PMID: 28379944]
[34]
Kakar SS, Sellers JC, Devor DC, Musgrove LC, Neill JD. Angiotensin II type-1 receptor subtype cDNAs: differential tissue expression and hormonal regulation. Biochemical and biophysical research communications. 1992 Mar 31:183(3):1090-6
[PubMed PMID: 1567388]
[35]
Sumners C, Alleyne A, Rodríguez V, Pioquinto DJ, Ludin JA, Kar S, Winder Z, Ortiz Y, Liu M, Krause EG, de Kloet AD. Brain angiotensin type-1 and type-2 receptors: cellular locations under normal and hypertensive conditions. Hypertension research : official journal of the Japanese Society of Hypertension. 2020 Apr:43(4):281-295. doi: 10.1038/s41440-019-0374-8. Epub 2019 Dec 18
[PubMed PMID: 31853042]
[36]
Iwanaga Y, Kihara Y, Takenaka H, Kita T. Down-regulation of cardiac apelin system in hypertrophied and failing hearts: Possible role of angiotensin II-angiotensin type 1 receptor system. Journal of molecular and cellular cardiology. 2006 Nov:41(5):798-806
[PubMed PMID: 16919293]
[37]
Allen AM, Zhuo J, Mendelsohn FA. Localization and function of angiotensin AT1 receptors. American journal of hypertension. 2000 Jan:13(1 Pt 2):31S-38S
[PubMed PMID: 10678286]
[38]
Eguchi S, Kawai T, Scalia R, Rizzo V. Understanding Angiotensin II Type 1 Receptor Signaling in Vascular Pathophysiology. Hypertension (Dallas, Tex. : 1979). 2018 May:71(5):804-810. doi: 10.1161/HYPERTENSIONAHA.118.10266. Epub 2018 Mar 26
[PubMed PMID: 29581215]
Level 3 (low-level) evidence
[39]
Kaschina E, Unger T. Angiotensin AT1/AT2 receptors: regulation, signalling and function. Blood pressure. 2003:12(2):70-88
[PubMed PMID: 12797627]
[40]
Naito T, Ma LJ, Yang H, Zuo Y, Tang Y, Han JY, Kon V, Fogo AB. Angiotensin type 2 receptor actions contribute to angiotensin type 1 receptor blocker effects on kidney fibrosis. American journal of physiology. Renal physiology. 2010 Mar:298(3):F683-91. doi: 10.1152/ajprenal.00503.2009. Epub 2009 Dec 30
[PubMed PMID: 20042458]
[41]
Billet S, Aguilar F, Baudry C, Clauser E. Role of angiotensin II AT1 receptor activation in cardiovascular diseases. Kidney international. 2008 Dec:74(11):1379-84. doi: 10.1038/ki.2008.358. Epub 2008 Jul 23
[PubMed PMID: 18650793]
[42]
Lazard D, Briend-Sutren MM, Villageois P, Mattei MG, Strosberg AD, Nahmias C. Molecular characterization and chromosome localization of a human angiotensin II AT2 receptor gene highly expressed in fetal tissues. Receptors & channels. 1994:2(4):271-80
[PubMed PMID: 7719706]
[43]
Ozono R, Wang ZQ, Moore AF, Inagami T, Siragy HM, Carey RM. Expression of the subtype 2 angiotensin (AT2) receptor protein in rat kidney. Hypertension (Dallas, Tex. : 1979). 1997 Nov:30(5):1238-46
[PubMed PMID: 9369282]
[44]
Tsutsumi K, Saavedra JM. Characterization and development of angiotensin II receptor subtypes (AT1 and AT2) in rat brain. The American journal of physiology. 1991 Jul:261(1 Pt 2):R209-16
[PubMed PMID: 1858948]
[45]
Wang ZQ, Moore AF, Ozono R, Siragy HM, Carey RM. Immunolocalization of subtype 2 angiotensin II (AT2) receptor protein in rat heart. Hypertension (Dallas, Tex. : 1979). 1998 Jul:32(1):78-83
[PubMed PMID: 9674641]
[46]
Namsolleck P, Recarti C, Foulquier S, Steckelings UM, Unger T. AT(2) receptor and tissue injury: therapeutic implications. Current hypertension reports. 2014 Feb:16(2):416. doi: 10.1007/s11906-013-0416-6. Epub
[PubMed PMID: 24414230]
[47]
Carey RM, Siragy HM. Newly recognized components of the renin-angiotensin system: potential roles in cardiovascular and renal regulation. Endocrine reviews. 2003 Jun:24(3):261-71
[PubMed PMID: 12788798]
[48]
Siragy HM, Inagami T, Ichiki T, Carey RM. Sustained hypersensitivity to angiotensin II and its mechanism in mice lacking the subtype-2 (AT2) angiotensin receptor. Proceedings of the National Academy of Sciences of the United States of America. 1999 May 25:96(11):6506-10
[PubMed PMID: 10339618]
[49]
Sampson AK, Moritz KM, Jones ES, Flower RL, Widdop RE, Denton KM. Enhanced angiotensin II type 2 receptor mechanisms mediate decreases in arterial pressure attributable to chronic low-dose angiotensin II in female rats. Hypertension (Dallas, Tex. : 1979). 2008 Oct:52(4):666-71. doi: 10.1161/HYPERTENSIONAHA.108.114058. Epub 2008 Aug 18
[PubMed PMID: 18711010]
[50]
Williams GH. Aldosterone biosynthesis, regulation, and classical mechanism of action. Heart failure reviews. 2005 Jan:10(1):7-13
[PubMed PMID: 15947886]
[51]
Quinn SJ, Williams GH. Regulation of aldosterone secretion. Annual review of physiology. 1988:50():409-26
[PubMed PMID: 3288099]
[52]
Arriza JL, Weinberger C, Cerelli G, Glaser TM, Handelin BL, Housman DE, Evans RM. Cloning of human mineralocorticoid receptor complementary DNA: structural and functional kinship with the glucocorticoid receptor. Science (New York, N.Y.). 1987 Jul 17:237(4812):268-75
[PubMed PMID: 3037703]
[53]
Holst JP, Soldin OP, Guo T, Soldin SJ. Steroid hormones: relevance and measurement in the clinical laboratory. Clinics in laboratory medicine. 2004 Mar:24(1):105-18
[PubMed PMID: 15157559]
[54]
McCormick JA, Bhalla V, Pao AC, Pearce D. SGK1: a rapid aldosterone-induced regulator of renal sodium reabsorption. Physiology (Bethesda, Md.). 2005 Apr:20():134-9
[PubMed PMID: 15772302]
[55]
Summa V, Mordasini D, Roger F, Bens M, Martin PY, Vandewalle A, Verrey F, Féraille E. Short term effect of aldosterone on Na,K-ATPase cell surface expression in kidney collecting duct cells. The Journal of biological chemistry. 2001 Dec 14:276(50):47087-93
[PubMed PMID: 11598118]
[56]
MacKenzie SM, Clark CJ, Fraser R, Gómez-Sánchez CE, Connell JM, Davies E. Expression of 11beta-hydroxylase and aldosterone synthase genes in the rat brain. Journal of molecular endocrinology. 2000 Jun:24(3):321-8
[PubMed PMID: 10828825]
[57]
Fuller PJ, Yao Y, Yang J, Young MJ. Mechanisms of ligand specificity of the mineralocorticoid receptor. The Journal of endocrinology. 2012 Apr:213(1):15-24. doi: 10.1530/JOE-11-0372. Epub 2011 Dec 12
[PubMed PMID: 22159507]
[58]
Geerling JC, Loewy AD. Aldosterone in the brain. American journal of physiology. Renal physiology. 2009 Sep:297(3):F559-76. doi: 10.1152/ajprenal.90399.2008. Epub 2009 Mar 4
[PubMed PMID: 19261742]
[59]
Xue B, Zhang Z, Roncari CF, Guo F, Johnson AK. Aldosterone acting through the central nervous system sensitizes angiotensin II-induced hypertension. Hypertension (Dallas, Tex. : 1979). 2012 Oct:60(4):1023-30. doi: 10.1161/HYPERTENSIONAHA.112.196576. Epub 2012 Sep 4
[PubMed PMID: 22949534]
[60]
Remuzzi G, Perico N, Macia M, Ruggenenti P. The role of renin-angiotensin-aldosterone system in the progression of chronic kidney disease. Kidney international. Supplement. 2005 Dec:(99):S57-65
[PubMed PMID: 16336578]
[61]
Orsborne C, Chaggar PS, Shaw SM, Williams SG. The renin-angiotensin-aldosterone system in heart failure for the non-specialist: the past, the present and the future. Postgraduate medical journal. 2017 Jan:93(1095):29-37. doi: 10.1136/postgradmedj-2016-134045. Epub 2016 Sep 26
[PubMed PMID: 27671772]
[62]
Schmieder RE, Hilgers KF, Schlaich MP, Schmidt BM. Renin-angiotensin system and cardiovascular risk. Lancet (London, England). 2007 Apr 7:369(9568):1208-19
[PubMed PMID: 17416265]
[63]
Manrique C, Lastra G, Gardner M, Sowers JR. The renin angiotensin aldosterone system in hypertension: roles of insulin resistance and oxidative stress. The Medical clinics of North America. 2009 May:93(3):569-82. doi: 10.1016/j.mcna.2009.02.014. Epub
[PubMed PMID: 19427492]
[64]
Ferrari R. RAAS inhibition and mortality in hypertension. Global cardiology science & practice. 2013:2013(3):269-78. doi: 10.5339/gcsp.2013.34. Epub 2013 Nov 1
[PubMed PMID: 24689028]
[65]
Rossi GP. Primary Aldosteronism: JACC State-of-the-Art Review. Journal of the American College of Cardiology. 2019 Dec 3:74(22):2799-2811. doi: 10.1016/j.jacc.2019.09.057. Epub
[PubMed PMID: 31779795]
[66]
Cohen JB, Cohen DL, Herman DS, Leppert JT, Byrd JB, Bhalla V. Testing for Primary Aldosteronism and Mineralocorticoid Receptor Antagonist Use Among U.S. Veterans : A Retrospective Cohort Study. Annals of internal medicine. 2021 Mar:174(3):289-297. doi: 10.7326/M20-4873. Epub 2020 Dec 29
[PubMed PMID: 33370170]
Level 2 (mid-level) evidence
[67]
Parving HH, Brenner BM, McMurray JJ, de Zeeuw D, Haffner SM, Solomon SD, Chaturvedi N, Persson F, Desai AS, Nicolaides M, Richard A, Xiang Z, Brunel P, Pfeffer MA, ALTITUDE Investigators. Cardiorenal end points in a trial of aliskiren for type 2 diabetes. The New England journal of medicine. 2012 Dec 6:367(23):2204-13. doi: 10.1056/NEJMoa1208799. Epub 2012 Nov 3
[PubMed PMID: 23121378]
[68]
Heerspink HJ, Persson F, Brenner BM, Chaturvedi N, Brunel P, McMurray JJ, Desai AS, Solomon SD, Pfeffer MA, Parving HH, de Zeeuw D. Renal outcomes with aliskiren in patients with type 2 diabetes: a prespecified secondary analysis of the ALTITUDE randomised controlled trial. The lancet. Diabetes & endocrinology. 2016 Apr:4(4):309-17. doi: 10.1016/S2213-8587(15)00469-6. Epub 2016 Jan 14
[PubMed PMID: 26774608]
Level 1 (high-level) evidence
[69]
Køber L, Torp-Pedersen C, Carlsen JE, Bagger H, Eliasen P, Lyngborg K, Videbaek J, Cole DS, Auclert L, Pauly NC. A clinical trial of the angiotensin-converting-enzyme inhibitor trandolapril in patients with left ventricular dysfunction after myocardial infarction. Trandolapril Cardiac Evaluation (TRACE) Study Group. The New England journal of medicine. 1995 Dec 21:333(25):1670-6
[PubMed PMID: 7477219]
[70]
Pfeffer MA, Braunwald E, Moyé LA, Basta L, Brown EJ Jr, Cuddy TE, Davis BR, Geltman EM, Goldman S, Flaker GC. Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction. Results of the survival and ventricular enlargement trial. The SAVE Investigators. The New England journal of medicine. 1992 Sep 3:327(10):669-77
[PubMed PMID: 1386652]
[71]
Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. The New England journal of medicine. 1993 Nov 11:329(20):1456-62
[PubMed PMID: 8413456]
[72]
Kamper AL, Strandgaard S, Leyssac PP. Effect of enalapril on the progression of chronic renal failure. A randomized controlled trial. American journal of hypertension. 1992 Jul:5(7):423-30
[PubMed PMID: 1637513]
Level 1 (high-level) evidence
[73]
Maschio G, Alberti D, Janin G, Locatelli F, Mann JF, Motolese M, Ponticelli C, Ritz E, Zucchelli P. Effect of the angiotensin-converting-enzyme inhibitor benazepril on the progression of chronic renal insufficiency. The Angiotensin-Converting-Enzyme Inhibition in Progressive Renal Insufficiency Study Group. The New England journal of medicine. 1996 Apr 11:334(15):939-45
[PubMed PMID: 8596594]
[74]
Cohn JN, Tognoni G, Valsartan Heart Failure Trial Investigators. A randomized trial of the angiotensin-receptor blocker valsartan in chronic heart failure. The New England journal of medicine. 2001 Dec 6:345(23):1667-75
[PubMed PMID: 11759645]
Level 1 (high-level) evidence
[75]
Pitt B, Segal R, Martinez FA, Meurers G, Cowley AJ, Thomas I, Deedwania PC, Ney DE, Snavely DB, Chang PI. Randomised trial of losartan versus captopril in patients over 65 with heart failure (Evaluation of Losartan in the Elderly Study, ELITE). Lancet (London, England). 1997 Mar 15:349(9054):747-52
[PubMed PMID: 9074572]
Level 1 (high-level) evidence
[76]
Young JB, Dunlap ME, Pfeffer MA, Probstfield JL, Cohen-Solal A, Dietz R, Granger CB, Hradec J, Kuch J, McKelvie RS, McMurray JJ, Michelson EL, Olofsson B, Ostergren J, Held P, Solomon SD, Yusuf S, Swedberg K, Candesartan in Heart failure Assessment of Reduction in Mortality and morbidity (CHARM) Investigators and Committees. Mortality and morbidity reduction with Candesartan in patients with chronic heart failure and left ventricular systolic dysfunction: results of the CHARM low-left ventricular ejection fraction trials. Circulation. 2004 Oct 26:110(17):2618-26
[PubMed PMID: 15492298]
[77]
Brenner BM, Cooper ME, de Zeeuw D, Keane WF, Mitch WE, Parving HH, Remuzzi G, Snapinn SM, Zhang Z, Shahinfar S, RENAAL Study Investigators. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. The New England journal of medicine. 2001 Sep 20:345(12):861-9
[PubMed PMID: 11565518]
[78]
Yoo TH, Hong SJ, Kim S, Shin S, Kim DK, Lee JP, Han SY, Lee S, Won JC, Kang YS, Park J, Han BG, Na KR, Hur KY, Kim YJ, Park S. The FimAsartaN proTeinuriA SusTaIned reduCtion in comparison with losartan in diabetic chronic kidney disease (FANTASTIC) trial. Hypertension research : official journal of the Japanese Society of Hypertension. 2022 Dec:45(12):2008-2017. doi: 10.1038/s41440-022-01028-6. Epub 2022 Sep 20
[PubMed PMID: 36123398]
[79]
Lewis EJ, Hunsicker LG, Clarke WR, Berl T, Pohl MA, Lewis JB, Ritz E, Atkins RC, Rohde R, Raz I, Collaborative Study Group. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. The New England journal of medicine. 2001 Sep 20:345(12):851-60
[PubMed PMID: 11565517]
[80]
Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A, Palensky J, Wittes J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. The New England journal of medicine. 1999 Sep 2:341(10):709-17
[PubMed PMID: 10471456]
Level 1 (high-level) evidence
[81]
Zannad F, McMurray JJ, Krum H, van Veldhuisen DJ, Swedberg K, Shi H, Vincent J, Pocock SJ, Pitt B, EMPHASIS-HF Study Group. Eplerenone in patients with systolic heart failure and mild symptoms. The New England journal of medicine. 2011 Jan 6:364(1):11-21. doi: 10.1056/NEJMoa1009492. Epub 2010 Nov 14
[PubMed PMID: 21073363]
[82]
Pitt B, Filippatos G, Agarwal R, Anker SD, Bakris GL, Rossing P, Joseph A, Kolkhof P, Nowack C, Schloemer P, Ruilope LM, FIGARO-DKD Investigators. Cardiovascular Events with Finerenone in Kidney Disease and Type 2 Diabetes. The New England journal of medicine. 2021 Dec 9:385(24):2252-2263. doi: 10.1056/NEJMoa2110956. Epub 2021 Aug 28
[PubMed PMID: 34449181]
[83]
Bakris GL, Agarwal R, Anker SD, Pitt B, Ruilope LM, Rossing P, Kolkhof P, Nowack C, Schloemer P, Joseph A, Filippatos G, FIDELIO-DKD Investigators. Effect of Finerenone on Chronic Kidney Disease Outcomes in Type 2 Diabetes. The New England journal of medicine. 2020 Dec 3:383(23):2219-2229. doi: 10.1056/NEJMoa2025845. Epub 2020 Oct 23
[PubMed PMID: 33264825]
[84]
Freeman MW, Halvorsen YD, Marshall W, Pater M, Isaacsohn J, Pearce C, Murphy B, Alp N, Srivastava A, Bhatt DL, Brown MJ, BrigHTN Investigators. Phase 2 Trial of Baxdrostat for Treatment-Resistant Hypertension. The New England journal of medicine. 2023 Feb 2:388(5):395-405. doi: 10.1056/NEJMoa2213169. Epub 2022 Nov 7
[PubMed PMID: 36342143]
[85]
Hricik DE, Dunn MJ. Angiotensin-converting enzyme inhibitor-induced renal failure: causes, consequences, and diagnostic uses. Journal of the American Society of Nephrology : JASN. 1990 Dec:1(6):845-58
[PubMed PMID: 2103846]
[86]
Murphy AM, Wong AL, Bezuhly M. Modulation of angiotensin II signaling in the prevention of fibrosis. Fibrogenesis & tissue repair. 2015:8():7. doi: 10.1186/s13069-015-0023-z. Epub 2015 Apr 23
[PubMed PMID: 25949522]
[87]
Nagai T, Nitta K, Kanasaki M, Koya D, Kanasaki K. The biological significance of angiotensin-converting enzyme inhibition to combat kidney fibrosis. Clinical and experimental nephrology. 2015 Feb:19(1):65-74. doi: 10.1007/s10157-014-1000-3. Epub 2014 Jul 1
[PubMed PMID: 24975544]