Continuing Education Activity
Prune belly syndrome is a rare congenitally acquired disorder primarily characterized by the clinical triad of deficient abdominal musculature, cryptorchidism, and urinary tract abnormalities. Children born with this syndrome present on a broad spectrum, ranging from incompatibility with life to aging normally and having children of their own. This activity reviews the evaluation and management of prune belly syndrome and highlights the role of the interprofessional team in managing patients with this complex condition.
Objectives:
- Identify the possible etiologies of prune belly syndrome.
- Describe the clinical and radiological findings associated with prune belly syndrome.
- Outline the management options available for prune belly syndrome.
- Summarize interprofessional team strategies for improving care coordination and communication among health care professionals managing prune belly syndrome and improving outcomes.
Introduction
Prune belly syndrome, also referred to as Eagle-Barrett syndrome or the triad syndrome, is a rare congenital disorder characterized by the triad of deficient abdominal musculature, cryptorchidism, and urinary tract abnormalities.[1] Children born with this condition present on a broad spectrum ranging from incompatibility with life to aging normally and having children of their own. The severity of renal dysplasia mostly determines the survival and prognosis among the survivors.[2] Perinatal mortality ranges between 10 to 25% in contemporary studies and directly correlates to the severity of pulmonary hypoplasia as a result of oligohydramnios from reduced fetal urine production from renal dysplasia and urinary tract abnormalities leading to Potter sequence.[3]
Etiology
The exact etiology of prune belly syndrome remains unknown, although there are several theories.[4][5][6][7] One theory is the urethral obstruction-malformation complex, and the proposal is that a urethral obstruction during embryological development produces bladder distension that has secondary effects on the development of the urinary tract, abdominal wall, and testicular descent.[7]A different theory ascribes this syndrome to a yolk sac defect.[8] Yet another theory points to a possible defect in the lateral plate mesoderm, which embryologically gives rise to the ureters, bladder, prostate, urethra, and gubernaculum.[6] No single explanation has universal acceptance, and some researchers believe in the combination of more than one theory in the causation. An underlying genetic abnormality is under investigation, but to date remains unproven.[9]
Epidemiology
Prune belly syndrome has a contemporary incidence of 3.6 to 3.8 per 100000 live male births.[10] It appears more predominantly in males, and less than 5% of those diagnosed are female.[2] Female patients will have the typical urinary tract findings and abdominal appearance characteristic of prune belly syndrome.[11]
Pathophysiology
Male infants with prune belly syndrome display bilateral cryptorchidism where the testes lie intra-abdominally, adjacent to the dilated ureters at the level of the iliac arteries. The reason for the undescended testes is not known.[2] Hydroureteronephrosis is almost always present and is mostly bilateral.[3]
The massive dilation of the ureters occur at the distal level; however, this finding is variable. Hydroureteronephrosis is seldom due to obstruction within the ureter; instead, the usual culprits include lower urinary tract obstruction (e.g., posterior urethral valves), vesicoureteral reflux (VUR) and a histologic deficiency of smooth muscle and preponderance of fibrous tissue in ureters leading to ineffective peristalsis.[12] Interestingly, in prune belly syndrome, there are more normal-appearing smooth muscle cells in the proximal ureter, which is essential to remember when planning and performing the ureteral reconstruction.[13] Around 75% of prune belly syndrome patients have VUR, and in most cases, it is bilateral.[3] Approximately one-half of the patients with prune belly syndrome will have renal dysplasia with variable severity, and about 40 to 50% will require renal replacement therapy at some point in time.[14] Favorable prognostic indicators include at least one normal-appearing kidney on ultrasound and a nadir serum creatinine of less than 0.7 mg/dL over their first year of life.[3][15]
The severity of abdominal wall musculature is variable. There are rare cases with completely absent abdominal wall musculature; the abdominal wall deficiency is often present in the lower part of the abdominal wall. The rectus muscles and internal and external obliques are often less well developed, and in severe cases, skin, subcutaneous fat and a single fibrous layer may be all that is overlying the peritoneum.
Besides the characteristic triad, about 75% of children with prune belly syndrome have abnormalities involving other systems.[16] These include respiratory (58%, e.g., pulmonary hypoplasia), cardiac (25%, e.g., patent ductus arteriosus, ventricular septal defect, atrial septal defect, and tetralogy of Fallot), gastrointestinal (24%, e.g., malrotation of the midgut, bowel atresia, anorectal anomalies) and musculoskeletal defects (22%, e.g., scoliosis, talipes equinovarus and hip dysplasia).[3][17]
History and Physical
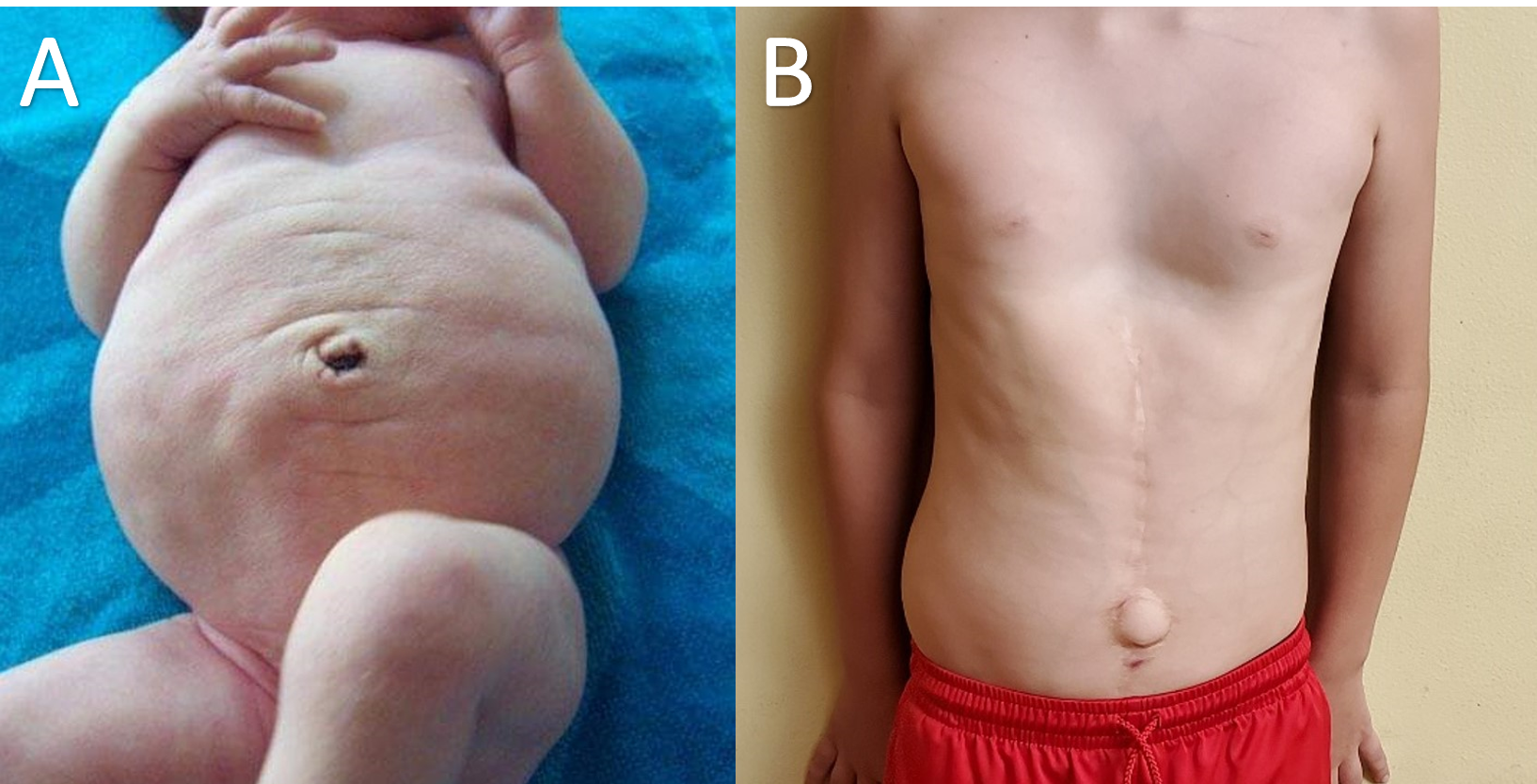
The characteristic dried prune-like wrinkled appearance of the abdominal wall is the most notable physical finding and is the reason for the name, prune belly syndrome. This abdominal finding appears to be attributable to either deficient or absent abdominal musculature.[17] However, as children with prune belly syndrome grow, the adipose tissue deposits in the subcutaneous layer of the abdominal wall, and this tends to diminish the wrinkled appearance. This change happens gradually over the first year of life when their abdomen begins to take on more of a pot-belly appearance owing to the deficiency of the underlying musculature. Interestingly, despite the deficiency in abdominal musculature, the gait is usually not affected, but the achievement of the motor milestones (e.g., walking) may experience a delay. Patients do, however, tend to sit up by rolling to their sides and using their arms to push themselves up from a supine position. Also, many patients may have lordosis.[18]
Evaluation
During the second trimester, suspected prune belly syndrome may be detectable during the prenatal ultrasound screening.[17] In severe cases, maternal oligohydramnios may present. Prenatal ultrasound may also indicate urinary tract abnormalities such as the distended bladder, dilated ureters, hydronephrosis, and also may demonstrate the deficient or absent abdominal muscle wall.[19]
In patients with severe renal dysplasia, pulmonary hypoplasia is a common finding. These patients are either stillborn or die in the early neonatal period. If alive at birth, these babies with pulmonary hypoplasia usually require aggressive neonatal resuscitation with various mechanical ventilation strategies. A chest radiograph is helpful to evaluate the pulmonary status, which generally shows small underdeveloped low volume lungs. A renal function panel should be ordered to assess renal status.
Once the neonate is stable, a renal and bladder ultrasound can take place, which helps evaluate the nature and severity of the urinary tract dysmorphism. Enlarged bladder with significant residual urine may be present due to weak bladder contraction.[17] An early voiding cystourethrogram (VCUG) is also indicated to assess for the presence of any VUR, but more importantly, to study the bladder outlet as well as the bladder emptying ability. Wide bladder neck and urethral malformations (including dilated prostatic urethra) are also commonly noted in VCUG.[20] Urodynamic study shows may indicate the poor detrusor contractility.[21] Echocardiogram of the heart and imaging (e.g., ultrasound and contrast radiographs) of the abdomen will help screen for the cardiac and gastrointestinal anomalies, respectively.
Treatment / Management
Medical: Nearly 80% of patients born with prune belly syndrome will have at least one documented urinary tract infection (UTI), and one-third of these patients will develop pyelonephritis.[3] The high likelihood of UTI owes mostly to the common comorbidities such as VUR, ureteral urinary stasis due to ineffective peristalsis and poor bladder emptying. Urologists commonly recommend prophylactic antibiotics and elective circumcision to minimize the risk of UTIs. Similarly, antibiotics are a necessity before any urinary tract manipulation, including patients undergoing a VCUG, as the risk of developing a UTI is high. Constipation is common in prune belly syndrome patients due to defective Valsalva maneuver required for defecation, and severe constipation can predispose to UTIs.[18] Similarly, defective abdominal muscles lead to an ineffective cough and predispose to respiratory infections requiring antibiotic therapies.[18] Patients with end-stage renal disease (ESRD) will require renal replacement therapy.
Surgical: Broadly, patients with PBS may undergo three types of surgeries: orchiopexy, procedures involving the urinary tract, and the abdominal wall reconstruction. The timing and sequence of these procedures vary among individuals based on the severity of the urinary tract abnormalities and also the presence of other comorbidities (e.g., pulmonary hypoplasia).
Orchiopexy is almost always an indicated procedure in prune belly syndrome, and the current recommendation for bilateral orchiopexy is at about six months of age. The orchiopexy at an earlier age is recommended but should be weighed against the higher anesthetic risks. A delayed orchiopexy worsens the testicular prognosis as in any other case of undescended testes.
The timing and need for urinary tract reconstructive surgeries vary among clinicians who manage prune belly syndrome, and the decision should be individually based on the clinical severity of each patient.[22] Some centers advocate early surgery with the belief that improvement of urinary stasis and VUR may improve the renal function.[22] In other places, a delayed approach with close surveillance for improvement in urinary tract obstruction and renal function is the recommended course.[22] Urinary tract reconstructive procedures are a strong recommendation in patients with recurrent febrile UTIs or with progressive renal deterioration. Similarly, abdominal wall reconstruction is necessary for children with moderate to severe abdominal wall deficiencies. Abdominoplasty may take place at any time but usually gets performed in coordination with other surgeries, including orchiopexy, vesicostomy, or urinary tract reconstruction. Apart from cosmetic reason, abdominoplasty procedures may improve the muscle tone, which may help in bladder emptying via an efficient Valsalva maneuver.[23]
Differential Diagnosis
Given the characteristic triad and the pathognomonic finding of the abdominal wall in prune belly syndrome, a differential diagnosis is often not required. It is crucial, however, to identify the severity and potential causes of the urinary tract dysmorphisms and also to consider all other comorbid conditions associated with prune belly syndrome outside of the usual characteristic triad. Some patients have the typical urinary tract abnormalities described in prune belly syndrome but may have either normally placed testes (or unilateral cryptorchidism) or a normal abdominal wall (or partial/unilateral laxity), referred to as pseudo-prune belly syndrome.[2][24] This pseudo-prune belly syndrome is identical to the megacystis-megaureter syndrome.[2]
Prognosis
As stated before, children born with this condition present on a spectrum ranging from incompatibility with life to leading a normal healthy life. Perinatal mortality ranges between 10 to 25% in contemporary studies and directly correlates to the level of prematurity and severity of pulmonary hypoplasia as a direct result of urinary tract abnormality leading to Potter sequence.[3] Additionally, about 40% are premature, and nearly half require mechanical ventilation at birth.[9] One-fourth of the patients also have congenital heart defects. These comorbidities also affect the outcome.
Woodard broadly classified prune belly syndrome patients into three clinical groups.[25] The first group (about 20% of all prune belly syndrome patients) characteristically demonstrates severe renal and pulmonary hypoplasia, and most babies are either stillborn or die shortly after birth.[25] The second group (about 40%) patients have an adequate renal function at birth.[25] Due to the significant renal abnormalities, the renal function may deteriorate later in life from ongoing urinary tract obstruction or frequent UTIs requiring aggressive management. The third group (about 40%) has the best prognosis with normal renal function despite mild urological abnormalities and lead near normal life.[25]
Before 1992, men with prune belly syndrome were considered infertile as there had been no reported cases of paternity.[26] Their infertility appeared to be multifactorial: mainly due to the cryptorchidism, and other factors including an incompetent bladder neck (leading to retrograde ejaculation), and prostatic hypoplasia. Currently, due to advancement in management options such as orchiopexy (done in a timely fashion as discussed above), sperm retrieval techniques, and intracytoplasmic sperm injection, many men with prune belly syndrome have successfully fathered children.
With only 5% of prune-belly syndrome diagnoses being female, females exhibit only deficiency of abdominal wall musculature and the anomalous urinary tract without any gonadal abnormality.[3] As such, there is a paucity in the literature regarding prune belly syndrome women, and there is minimal information regarding their fertility prospects. There is a report of a normal pregnancy with vaginal birth in a woman with prune belly syndrome.[27]
Complications
Given the multi-organ system manifestations, many potential complications abound. The most important to keep in mind, however, are the ones which clinicians can significantly impact the course and improve the outcome.
- In severe cases, the complications of pulmonary hypoplasia and renal dysplasia predominate. These require management by the multidisciplinary team, including the maternal-fetal medicine specialists, neonatologists, urologists, nephrologists, geneticists, pulmonologists, anesthesiologists, respiratory therapists, and others. Families should be thoroughly informed about the condition of the fetus (or infant), and receive counseling about the possible outcome. They should be helped to make informed decisions, and all the ethical considerations should be addressed by the team of experienced specialists.
- Beyond the neonatal period, urinary tract complications predominate, and about one-third of patients may eventually progress to need a renal transplant.[17] In a study involving 35 prune belly syndrome patients, the presence of bilateral abnormal kidneys on imaging, a nadir creatinine over 0.7 mg/dL, and pyelonephritis were poor prognostic factors.[15] It is essential to make an early diagnosis, especially in milder cases if the classic triad is not present. Urological consultation for a thorough evaluation of the urinary tract obstruction and prevention of further complications is usually necessary (e.g., antibiotic prophylaxis/circumcision should be planned to prevent UTIs).
Deterrence and Patient Education
- As there is no known cause for prune belly syndrome, there are no steps for the prevention of this syndrome.
Pearls and Other Issues
- Babies born with prune belly syndrome present on a spectrum ranging from incompatibility with life to leading a normal healthy life. The care plan requires individualization based on the type and severity of the presentation.
Enhancing Healthcare Team Outcomes
As prune belly syndrome has manifestations in nearly every major organ system, it truly is a diagnosis that requires a large team of specialists. The role of the neonatologist or pediatrician is vital to appropriately navigate the early management and workup of a child born with prune belly syndrome. Pediatric surgery and urology consults are important initially to manage the classic triad of symptoms. Other specialists should be consulted (cardiology, orthopedic surgery, pulmonology, gastroenterology, pediatric surgery, cardiac surgery) depending upon where the work-up leads.
As prune belly syndrome is a condition with variable severity and with multisystemic manifestations, patients are best managed by the interprofessional team. In severe cases with poor outcome, families should be thoroughly informed about the condition of the infant (or fetus), and appropriate, timely counseling should be provided. They should be helped to make informed decisions, and all the ethical considerations should be addressed by the team of experienced specialists.
In patients with less severe manifestations, it is of utmost importance to educate and support the parents of infants born with prune belly syndrome through the early perinatal period and ensure that they understand the importance of continued follow-up and surgical planning. Many parents will be relieved to know that the prune belly syndrome will improve over time, not every patient needs complex reconstructive surgeries and that their child will potentially live a normal life with the ability to have kids of their own in the future should they so desire.
For parents, it is essential to educate them that orchiopexy should be done near the six months age mark, and this time frame is also an appropriate period to plan an abdominal wall reconstructive surgery, if necessary. Urinary tract reconstruction is usually avoided unless they develop recurrent febrile urinary tract infections or demonstrate progressive renal deterioration. Occasionally children will need temporizing urologic interventions such as a cutaneous vesicostomy in order to safely drain the bladder while infants grow in preparation for more definitive surgeries such as ureteral reconstruction and reimplantation at a later age. Clinicians can refer families with PBS to peer support groups for further resources such as Prune Belly Syndrome Network.
Prune belly syndrome patients often require surgical procedures and are at a higher risk of anesthetic complications because the deficiency in abdominal wall musculature, which compromises their cough effectiveness, potentially leading to retention of pulmonary secretions and subsequent pneumonia. During hospitalizations, they should also be closely watched for the development of atelectasis for the same reason.
Whenever catheterization is indicated, whether for voiding cystourethrogram or obstruction of the bladder outlet necessitating foley drainage, nursing plays a key role in assuring that proper sterile technique is utilized during catheter placement and that proper catheter hygiene is continued with the foley remains in place.
Patients with prune belly syndrome do appear to be at a higher risk for testicular malignancy, but it is no higher than that of non-prune belly syndrome patients with cryptorchidism. This is one more reason that placement of the testes into the scrotum in a timely manner is necessary to both reduce the risk of developing malignancy as well as enhancing the detection of potential tumors.
The only way to improve outcomes is by managing these patients with an interprofessional team with close communication. Because of the multisystem organ involvement, most patients need lifelong follow up by the pediatrician, nurse practitioner, social worker, urologist, and the primary care provider.
Each of these clinicians has a particular role and contribution to make the overall team. Physicians will manage the case overall, and decide which specialists, as outlined above, will be necessary. Nursing will coordinate ongoing care and can offer counsel and direction to parents, and in surgical cases, they are on the front lines for post-surgical care. If antibiotics are necessary, the pharmacist should verify all dosing and assess coverage. Social workers or counselors should provide reassurance and ongoing emotional support. In each of these roles, the individual contributor must report back to the team and document progress within their specialty. Only through this type of collaboration can the interprofessional team drive outcomes in prune belly syndrome to successful outcomes, regardless of the severity of the particular case. [Level V]