Continuing Education Activity
Ornithine transcarbamylase deficiency (OTCD) is the most prevalent genetic disorder within urea cycle disorders, characterized by X-linked inheritance and variable phenotypic expression. Homozygous males with severe neonatal onset present shortly after birth with hyperammonemia, while late-onset cases may display mild neurocognitive symptoms. The severity of OTCD ranges from potentially lethal to enduring neurocognitive deficits. Interdisciplinary healthcare management involving clinical providers, nurses, pharmacists, and dieticians significantly enhances patient outcomes. Although genetic testing is often confirmatory, recent challenges in diagnosis highlight the importance of clinical and biochemical suspicion.
This activity reviews the comprehensive pathophysiology of OTCD, clinical presentation, and diagnostic evaluation, emphasizing the critical role of a coordinated interprofessional approach in optimizing clinical outcomes and reducing associated morbidity and mortality.
Objectives:
Identify the typical clinical presentation of ornithine transcarbamylase deficiency in newborns.
Implement an appropriate management plan for treating hyperammonemia in patients with ornithine transcarbamylase deficiency.
Develop a long-term clinical management plan for patients with ornithine transcarbamylase deficiency.
Collaborate with an interprofessional team of clinicians, dieticians, and pharmacists in managing ornithine transcarbamylase deficiency to help improve clinical outcomes for patients affected by this disease.
Introduction
Ornithine transcarbamylase deficiency (OTCD) is a hereditary metabolic disorder and the most common urea cycle disorder (UCD).[1] This X-linked genetic condition affects ureagenesis, displaying variable phenotypic expressions. Homozygous males with severe neonatal onset typically exhibit symptoms such as hyperammonemia, respiratory alkalosis, and hypothermia.[1][2] Additionally, late-onset manifestations can occur in males with partial deficiency and in heterozygous females.[2]
In suspected individuals, genetic testing is usually confirmatory; however, studies have documented instances where routine Sanger sequencing of the ornithine transcarbamylase (OTC) gene did not identify any disease-causing variants.[3] Furthermore, next-generation sequencing and UCD gene panels proved unhelpful in diagnosing these cases. This underscores the importance of clinical and biochemical suspicion in diagnosing this rare yet potentially devastating disorder.[3]
This activity comprehensively examines the pathophysiology, clinical presentation, and diagnostic evaluation of OTCD. Emphasizing the pivotal role of an interprofessional team underscores the importance of collaborative management to optimize clinical outcomes and reduce the morbidity and mortality associated with this disease.
Etiology
OTCD is an X-linked defect in ureagenesis and is the most prevalent type of UCD globally.[1] The OTC enzyme, denoted as EC 2.1.3.3, is encoded by the OTC gene located on the short arm of the X chromosome at Xp11.4. The OTC gene exhibits numerous disease-causing mutations, yet not all individuals diagnosed with OTCD (based on reduced enzyme activity) have an identifiable pathogenic variant. Approximately 20% of patients diagnosed with OTCD fall in this category and are presumed to have variants in the regulatory or deep intronic regions.[1]
The disorder exhibits significant phenotypic heterogeneity, with clinical severity varying according to the type of mutation, genetic background, and environmental factors.[1] A recent study conducted in Japan identified 523 genetic variants causing OTCD,[4] encompassing 330 missense, 53 nonsense, and 3 silent substitutions, and 8 deletions, 2 duplications, 1 deletion insertion, 55 frameshifts, 2 extensions, and 69 "no category" mutations. The "no category" mutations included regulatory and splice site errors. The study highlighted that amino acid substitutions at the same position could cause neonatal or late-onset OTCD. The authors noted that the time of disease onset varied based on environmental factors.[4]
These environmental factors typically include prolonged fasting, a high-protein diet, pregnancy, and surgery.[5] However, patients with OTCD may be at risk for recurrent hyperammonemic crises due to various other conditions. Potential triggers for hyperammonemic crises in these individuals include infections, vomiting, gastrointestinal or internal bleeding, decreased energy or protein intake, catabolism and involution of the uterus during the postpartum period, chemotherapy, high-dose glucocorticoids, and unusual protein load leading to a significant increase in protein intake from baseline.[6] Additionally, certain medications have been identified as potential triggers for hyperammonemic episodes, including valproate, L-asparaginase/pegaspargase, topiramate, carbamazepine, phenobarbitone, phenytoin, primidone, furosemide, hydrochlorothiazide, and salicylates.[6]
The risk of symptomatic disease in female heterozygotes is contingent upon differential X chromosome inactivation. Specifically, the likelihood of symptoms in heterozygote females is higher if an affected male in the family experiences neonatal-onset disease compared to females with a male relative who has late-onset disease.[2]
Epidemiology
The estimated prevalence of OTCD is in the range of 1 in 14,000 to 1 in 80,000.[1] Based on newborn screening data, incidence rates have been reported at approximately 1 in 62,000 in Finland, 1 in 63,000 in the US, and 1 in 69,904 in Italy.[1]
As previously stated, OTCD is the most common UCD.[7] UCDs manifest in 1 of 8200 live births in the US,[8] rendering them more common within the US than globally.[9] OTCD is more frequently observed in neonates and early childhood than in adulthood.[10] Males are more prone to experiencing severe symptoms during the neonatal period due to the X-linked nature of the mutation.
Based on previous data, approximately 10% of female carriers exhibit symptoms.[11] Recent studies highlight that these estimates may not account for individuals with subtle symptoms that might not prompt medical attention, suggesting that the reported figures likely underestimate the prevalence of OTCD symptoms in female heterozygotes.[12]
In a recent study examining the epidemiology of OTCD in France, Turkey, and the United Kingdom (UK), findings revealed that in the French cohort, 34% were males, 25% were asymptomatic, and 41% were symptomatic females.[1] In Turkey, 38% were males, 16% were asymptomatic, and 46% were symptomatic females. In the UK, 33% were male, 41% were asymptomatic, and 26% were symptomatic females. The study further indicated that neonatal-onset cases accounted for 19%, 12%, and 7% in France, Turkey, and the UK, respectively. Late-onset were reported at 39%, 42%, and 67%, respectively.[1] Notably, UCDs are not included in routine neonatal screening in these countries, likely underestimating the actual number of cases.
OTCD is primarily an inherited disease, with 36% to 60% of patients indicating a positive family history.[1] While neonatal-onset OTCD historically constituted around 60% of all cases, recent data suggests that only 18% of symptomatic patients experienced neonatal onset, with the majority (82%) exhibiting post-neonatal onset. This discrepancy is likely due to the underrepresentation of individuals with mild symptoms in older data. Additionally, individuals with severe neonatal onset who die before enrollment may be underrepresented in newer studies.[2]
Pathophysiology
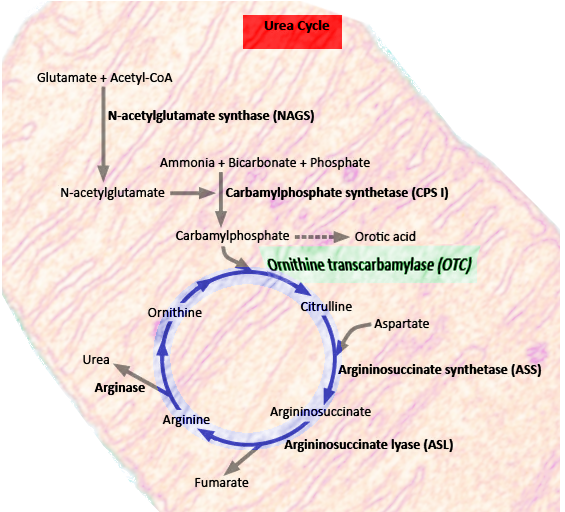
OTC is a critical enzyme in the urea cycle, a biochemical pathway responsible for converting nitrogenous waste into water-soluble urea (see Image. Urea Cycle). This process facilitates the elimination of nitrogenous waste products through urination. Any dysfunction in the urea cycle can result in hyperammonemia and resultant complications.
OTC (EC 2.1.3.3) is an evolutionarily "ancient" enzyme in bacteria, plants, and other vertebrates.[5] Its primary function involves facilitating the transfer of the carbamoyl group from carbamoyl phosphate to the amino group of L-ornithine, producing citrulline and phosphate. This enzymatic conversion plays a crucial role in maintaining ammonia homeostasis, the urea cycle, and the biosynthesis of citrulline.[5] Additionally, OTC is essential for arginine biosynthesis.[13]
In mammals, OTC is a mitochondrial protein expressed predominantly in the liver and intestine. In the liver, OTC plays a crucial role in the urea cycle by capturing nitrogen from ammonia to generate urea, facilitating excretion, and preventing ammonia buildup. Citrulline, a byproduct generated in the liver, contributes to the urea cycle and is not utilized systemically. However, citrulline, produced by the OTC enzyme in intestinal epithelial cells, is involved in amino acid biosynthesis.[5]
The tissue-specific regulation of OTC is controlled by a 5' promoter and an enhancer located upstream of the transcription start site. Liver-selective transcription factors, such as hepatocyte nuclear factor 4 (HNF-4) and CCAAT/enhancer-binding protein beta, activate the OTC enhancer.[5] The hepatic expression of this enzyme is subject to regulation based on nutritional states, with evidence from animal models indicating that high protein intake can lead to an increase in OTC expression.[5]
A comprehensive overview of disease-causing mutations in OTC reveals 538 described mutations. Among these, 28.81% are loss of function mutations (frameshifts, deletions, duplications, nonsense mutations, and other complex mutations), 57.06% affect the sequence of the protein (missense, inframe deletions, or inframe insertions), 11.52% involve splice sites, 1.49% affect the promotor, 0.74% causing loss of start codons, and 0.37% cause loss of STOP codons.[5]
OTCD follows an X-linked inheritance pattern, but de novo variants may also exist.[12] Males carrying a hemizygous disease-causing variant may experience severe life-threatening neonatal disease. Females, as heterozygous, and males with less severe variants can exhibit variable phenotypes, depending upon the degree of enzyme deficiency. In certain females, the disease manifestation may be severe if the X chromosome carrying the pathogenic allele is active.[12]
OTCD presentations are not uniform, even with the same pathogenic variant. A case study highlighted this clinical heterogeneity within 3 females of the same family carrying identical pathogenic variants, each exhibiting distinct clinical presentations.[12] Notably, the study involved 2 monozygotic twins, where one succumbed at a young age to acute liver failure and hyperammonemia following an acute viral illness, while the other twin remained clinically asymptomatic despite experiencing the same viral illness. Despite sharing the same pathogenic variant and experiencing the same environmental trigger, the variability in symptoms among these patients is attributed to differential X chromosome inactivation in hepatocytes.[12]
Late-onset OTCD in males has been associated with the c.-106C>A variant that occurs in the promoter region of the OTC gene. This variant has been demonstrated to impair promoter activity, restricting peak enzyme activity during periods of high metabolic stress owing to the defective binding of gene transcription factors. The variant remains clinically undetected in the absence of elevated metabolic stress.[3]
History and Physical
OTC deficiency is not included in routine neonatal screening in most centers. Patients are often identified either when symptoms manifest or through genetic testing following the detection of an affected problem in the family. Neonatal signs of OTCD include tachypnea, vomiting, and lethargy, usually emerging within the first few days of life.[14] In the late-onset variant, symptoms are nonspecific and highly variable, including lethargy, loss of appetite, early morning headaches, and confusion.[10] Poorly managed OTCD can result in behavioral and neurological symptoms, including seizures, aggressive behavior, encephalopathy, coma, and death.[15]
In an epidemiologic study conducted in Europe, altered neurological status and encephalopathy were the most common presenting symptoms in patients with OTCD.[1] Vomiting was identified as the predominant gastrointestinal symptom, with some patients reporting liver failure or elevated liver transaminases as their presenting symptom.[1]
Neonatal Onset
Infants with severe OTCD typically appear asymptomatic at birth but develop symptoms related to hyperammonemia within 2 to 3 days of life. Initial signs include reduced intake and hypotonia, which progresses to lethargy and somnolence. Hyperventilation and seizures, often subclinical and detected through electroencephalographic monitoring, are commonly observed. Critically ill neonates may present with additional features, such as hypothermia, respiratory alkalosis, and severe encephalopathy.[2]
Postneonatal Onset
In postneonatal presentations of infants, symptoms may arise when transitioning from breast milk to formula or whole milk. These infants can exhibit episodic vomiting, irritability, failure to thrive, and developmental delay.[2]
Older children or adults with late-onset OTCD can present with erratic behavior and delirium, typically preceded by physiologic or psychologic stressors, such as significant injury or surgery, severe illness, high-protein intake, prolonged fasting, postpartum condition, or cancer treatment.[2]
Heterozygous Females
Heterozygous females may either be asymptomatic or symptomatic with recurrent hyperammonemia. Those with symptomatic disease may self-impose protein intake restrictions and might go undiagnosed. Some heterozygous females may exhibit only mild cognitive deficits in executive functioning and motor capability without developing hyperammonemia or experiencing altered mental status.[2]
Evaluation
When available, newborn screening (NBS) for OTCD relies on quantifying citrulline levels on dried blood spots. Supplementary testing involving a glutamine-to-glutamate ratio is recommended to identify individuals with a UCD.[2] Low citrulline levels and elevated glutamine levels on NBS are indicative of OTCD. However, due to the inherent instability of glutamine and the limited specificity and sensitivity of citrulline for OTCD, current consensus guidelines do not recommend routine NBS for OTCD.[6]
Following abnormal citrulline results, further investigation involves assessing plasma ammonia levels, amino acid levels, urine organic acid levels, and urine orotic acid quantification.[2] A high glutamine concentration and a low citrulline concentration can also be observed in other proximal UCDs, such as N-acetyl glutamate synthetase (NAGS) and carbamoyl phosphate synthetase I (CPSI) deficiency.[2] Differentiating OTCD from these conditions is often accomplished by examining urinary orotic acid concentration, a key parameter in most diagnostic algorithms.
According to the diagnostic criteria established by the Longitudinal Study of Urea Cycle Disorders, confirmation of OTCD in an individual requires suggestive clinical and biochemical findings (such as elevated ammonia, elevated glutamine, and low-to-normal citrulline levels), the absence of another inborn error of metabolism, and 1 of the following:[2]
- A pathogenic (or likely pathogenic) variant in the OTC gene by molecular genetic testing.
- Increased orotic acid excretion of greater than or equal to 20 µmoles/mmol of creatinine after an allopurinol challenge or in a random urine collection.
- Decreased OTC enzyme activity in the liver of a male proband using a liver biopsy specimen.
Notably, liver biopsy is not recommended to establish the diagnosis in females, as differential X chromosome inactivation may lead to false-negative results. This emphasizes the importance of molecular genetic testing in females for a more accurate and conclusive diagnosis of OTCD.
Molecular testing begins with a single-gene analysis of the OTC gene, aiming to identify small intragenic deletions/insertions, missense, nonsense, and splice site variants. In cases where no variant is detected through sequencing, subsequent gene-targeted deletion/duplication analysis is undertaken to detect exon and whole-gene deletions or duplications. This combined approach, involving both sequence analysis and deletion/duplication analysis, reveals a defect in 80% to 90% of affected individuals (biochemically confirmed OTCD patients).[2] Genetic testing for OTCD confirmation may involve samples from liver tissue, intestinal mucosa, erythrocytes, and fibroblasts.[6]
The gold standard for diagnosing OTCD is using a liver biopsy specimen to measure OTC enzyme activity. This diagnostic approach is particularly valuable for individuals who do not test positive for any pathogenic OTC variants but exhibit a high clinical suspicion of OTCD. In cases of severe OTCD deficiency, the enzyme activity is typically less than 20%, but it may be as high as 30% in milder cases.[2] Due to differential X chromosome inactivation, enzyme activity analysis on liver biopsy specimens in heterozygote females may not represent the total OTC activity.[2]
Biochemical Abnormalities in OTCD
Understanding the biochemical profile of OTCD is crucial, especially in diagnosing patients without identifiable genetic variants. Hyperammonemia, a hallmark abnormality in UCDs, is consistently elevated in individuals with OTCD. While a normal ammonia level in a symptomatic neonate virtually excludes UCDs, this is not necessarily the case in adults.[6]
The following steps involve measuring serum glutamine and citrulline levels upon establishing hyperammonemia. In OTCD, glutamine levels will be elevated, while citrulline levels will be decreased. Following this, urinary orotic acid measurement is essential, which will be elevated in individuals with OTCD. Ornithine levels will typically remain normal in OTCD, accompanied by decreased serum arginine levels and increased alanine levels. According to the consensus statement from European guidelines, the diagnostic criteria for OtCD involve a combination of hyperammonemia with low serum citrulline levels, low serum arginine levels, and elevated urinary orotic acid levels.[6]
Ornithine aminotransferase (OAT) deficiency and hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome share a similar biochemical profile, exhibiting elevated serum glutamine levels, decreased serum citrulline levels, and elevated urinary orotic acid levels. However, a distinguishing factor is that serum ornithine levels will be elevated in these disorders. Furthermore, HHH is differentiated by elevated urinary homocitrulline levels.[6]
It is important to remember that while biochemical tests provide valuable insights, they are not the preferred method of confirming the diagnosis when genetic testing is available. Molecular genetic analysis augmented by array-comparative genomic hybridization, RNA-based sequencing, and multiplex ligation-dependent probe amplification (MLPA) methods is recommended to increase the sensitivity.[6] Enzymatic biochemical analysis remains beneficial in the small percentage of patients for whom genetic testing is unrevealing.
Treatment / Management
The treatment for OTCD involves a comprehensive and multidisciplinary approach to manage the condition and prevent hyperammonemic crises. Management can be broken down into the acute and long-term phases.
Acute Management
During the acute phase, rapid correction of hyperammonemia to less than or equal to 200 µmol/L is recommended. Consensus guidelines for managing hyperammonemia in pediatric patients advocate for key measures, including ensuring adequate hydration, discontinuing protein intake, and reversing catabolism as primary goals in acute management.[16]
Rehydration with a dextrose-containing fluid at a high infusion rate is recommended to reverse the catabolic state. However, careful glucose monitoring is essential to prevent hyperglycemia, which can lead to dangerous hyperosmolarity and should be avoided.[16] In some cases, insulin may be required to prevent severe hyperglycemia. According to some expert guidelines, if severe hyperglycemia with an elevated lactate level (>3 mmol/L) develops, reducing the glucose infusion rate is preferred over increasing insulin to address hyperglycemia.[6]
To minimize ammonia production, temporarily ceasing protein intake is advised. In cases of mild hyperammonemia (at the upper limit of normal for their age), stopping protein intake and initiating intravenous glucose and lipids formulation may suffice. However, ensuring adequate caloric intake through glucose and lipid infusions is crucial. Protein intake must be reintroduced within a maximum of 48 hours, ideally following the normalization of the ammonia level. Limiting protein for longer durations is not recommended as it will worsen protein catabolism and hyperammonemia.[16] Some guidelines recommend protein intake to be withheld for a maximum of 24 hours.[6]
Treatment of Hyperammonemia
Nitrogen scavengers such as sodium benzoate, sodium phenylacetate, sodium phenylbutyrate, and glycerol phenylbutyrate are used to offer alternative routes for nitrogen excretion. The therapy holds the advantage of rapid initiation compared to renal replacement therapy.[16] In the acute setting, intravenous formulations of sodium phenylacetate and sodium benzoate are recommended. Oral formulations containing sodium phenylbutyrate or sodium benzoate are utilized for long-term maintenance therapy.[2] Intravenous sodium phenylacetate and sodium benzoate function by "scavenging" ammonia, creating an alternate pathway for nitrogen excretion.[17] Careful monitoring for potential toxicity of these agents is necessary, as adverse effects such as nausea, vomiting, metabolic acidosis, and changes in mentation may occur.[18] Parenteral nitrogen scavenger therapy, combined with adequate caloric intake to prevent catabolism, has improved survival outcomes in patients experiencing hyperammonemic crises.[19] Notably, hypernatremia is a common complication of this therapy, and when parenteral nitrogen scavengers are administered, efforts should be made to minimize sodium from other infusion fluids.
Hemodialysis may be necessary to control hyperammonemia, particularly in cases of severely elevated ammonia levels or if there is no response to intravenous sodium phenylacetate and sodium benzoate therapy. Hemodialysis is the most effective way of rapidly eliminating excess ammonia. An emergent need for hemodialysis is warranted if the serum ammonia level exceeds 500 µmol/L. Dialysis may be considered for lower levels if there is an inadequate clinical response after 4 hours of medical management. A serum ammonia level above 300 µmol/L will probably require hemodialysis for proper correction and should be planned accordingly with the initiation of parenteral nitrogen scavenger therapy in the interim. If the ammonia level falls between 100 to 300 µmol/L and the patient exhibits severe encephalopathy or seizures with a poor response to nitrogen scavengers, dialysis should be strongly considered.[16]
Continuous venovenous hemodialysis (CVVHD) is considered superior to conventional hemodialysis (HD) and peritoneal dialysis (PD) in managing hyperammonemia. While all forms of continuous renal replacement therapy (CRRT) are deemed safe and effective, CVVHD has demonstrated higher ammonia clearance rates than CRRT. As a result, high-dose CVVHD is recommended as the first-line treatment for hyperammonemia in these patients.[16] For hemodynamically unstable neonates, hybrid therapy combining hemodialysis or CRRT with extracorporeal membrane oxygenation (ECMO) is recommended, as it increases the patient's blood volume and facilitates using a larger cannula.[16] However, exchange transfusion is not recommended for this purpose.[6]
Intravenous arginine hydrochloride is used to manage hyperammonemia and metabolic decompensation in individuals with OTCD to replace arginine, typically synthesized using a functional OTC enzyme. In OTCD, arginine becomes an essential amino acid, and its deficiency results in a catabolic state of protein breakdown, further mobilizing nitrogen. Arginine is crucial for generating water-soluble urea cycle intermediates, such as citrulline and argininosuccinic acid, which can form and be excreted when supplemental arginine is provided to these patients.[20] Careful hemodynamic monitoring is imperative during arginine supplementation, as it can lead to hypotension.[16]
Optimizing Caloric Intake in the Acute Phase
Optimal caloric intake is essential in reversing the catabolism present in patients experiencing hyperammonemic crises due to OTCD. To reverse catabolism, the recommended caloric intake is at least 120 to 130 kcal/kg/d.[21] Initially, this caloric intake should be protein-free (consisting of dextrose and lipids). When reintroducing protein, it is recommended to commence at 0.6 g/kg/day, administered as essential amino acids for 24 to 48 hours. If well-tolerated, the protein concentration can gradually increase to 1.2 g/kg/day, with half provided as essential amino acids and the other half via natural protein sources such as infant formula or breast milk. It is advisable to avoid elemental formulas, particularly those high in nitrogen when using infant formulas. If the patient continues to respond positively, protein intake can gradually increase to a maximum of 2 g/kg/d.[21]
Long-Term Management
Long-term management necessitates strict protein control and sufficient protein and caloric intake to facilitate growth. In some cases, an essential amino acid medical food may be needed to maintain normal essential amino acid levels.[2] To address nutritional needs, vitamins and trace minerals should be supplemented in a calorie-rich, protein-free formula or as an individual supplement. The primary goal in long-term management is to prevent recurrent hyperammonemia and ensure adequate development. This is achieved by ensuring that the patient adheres to the following components:[6]
- A low-protein diet
- Adequate supplementation of citrulline and arginine
- Adequate supplementation of essential amino acids, vitamins, and minerals
- Sufficient doses of nitrogen scavenger therapies.
A comprehensive treatment plan should also be established to address potential triggers for hyperammonia, such as infections and surgeries. This proactive approach aims to prevent recurrent hyperammonemic episodes and ensures a well-managed response to specific situations.[6]
Implementing a low-protein diet minimizes the patient's exogenous nitrogen load, and the optimal protein load is determined individually through a gradual titration of dietary protein against serum ammonia levels. Once the patient tolerates an oral diet and maintains a plasma ammonia level below 100 µmol/L, the transition to oral forms of nitrogen scavenger medications and arginine may be considered.
Optimizing protein intake to meet the recommended daily allowance for age is crucial, provided the patient can tolerate the load without exacerbating serum ammonia levels. This represents a pivotal aspect of long-term management, enabling normal growth and development. Many patients, particularly males, may require chronic nitrogen scavenger therapy to facilitate adequate protein intake. However, chronic use of nitrogen scavenger therapy, especially sodium phenylbutyrate, can lead to branched-chain amino acid deficiencies. Addressing this deficiency necessitates higher protein allowance, potentially worsening ammonia levels. Properly balancing between natural protein prescription and amino acid supplementation can help mitigate this challenge. A recent multicenter study evaluating UCDs reported that the use of amino acid mixtures supplemented with branched-chain amino acids (L-valine, L-isoleucine, and L-leucine) allowed for a lower natural protein prescription, maintaining adequate serum levels of branched-chain amino acids for these patients.[22]
Patients with OTCD may be susceptible to deficiencies in essential fatty acids and long-chain polyunsaturated fatty acids. This deficiency can be addressed through supplementation with essential fatty acid-enriched infant formulas or oils rich in polyunsaturated fatty acids. Vitamin and trace element supplementation is usually required to ensure comprehensive nutritional support for individuals with OTCD.[6]
Chronic supplementation with L-citrulline or L-arginine is essential for maintaining adequate arginine levels and preventing catabolism in patients with OTCD. The bioavailability of L-citrulline surpasses that of L-arginine, and studies have demonstrated that citrulline supplementation achieves higher plasma arginine levels in individuals with UCDs than those supplemented with arginine alone.[22] Recent data also indicates that decompensation episodes in patients receiving arginine monotherapy were associated with higher plasma ammonia levels compared to those receiving citrulline monotherapy or a combination of citrulline and arginine therapy.[23] A retrospective study from Japan also suggests that L-citrulline can reduce plasma ammonia levels and should be considered standard therapy in OTCD patients.[24]
Adherence to dietary recommendations, nitrogen scavenger medications, and citrulline (with or without arginine supplementation) has positively impacted long-term clinical outcomes. Studies reported no acute metabolic decompensations after the initial diagnostic episode in over 54% of the patients who maintained compliance.[23] While a higher incidence of decompensation episodes was noted in patients receiving a combination of citrulline plus arginine therapy in one study, the authors concluded that this reflected the underlying severity of their disease.
For long-term management, it is advisable to monitor glutamine concentrations as elevated levels may indicate poor metabolic control and an increased risk of recurrent hyperammonemia. Biochemical monitoring goals include achieving a target ammonia level of less than 80 µmol/L, a glutamine level of less than 1000 µmol/L, ensuring arginine is within the high normal range, and maintaining essential amino acids as well as branched-chain amino acids within the normal range.[6]
Liver transplantation is the most effective treatment for patients with severe neonatal-onset OTCD, effectively preventing recurrent hyperammonemic episodes and deterring further neurodevelopmental deterioration.[2] Although the procedure is curative, it has historically been associated with complications, particularly in pediatric populations, limiting its application. However, recent data from a tertiary care hospital in Spain indicates advancements in transplantation techniques, suggesting that liver transplantation is a viable and beneficial option for patients with OTCD. Early consideration of liver transplantation can minimize and, in some cases, completely prevent cognitive impairment, with notable post-transplant nutritional and developmental "catch-up," particularly in those undergoing early transplantation.[25]
Emerging molecular treatments are currently being explored. Engineered adeno-associated viral (AAV) vectors have shown promise in animal models as a potential option for treating this disease.[26]
Liver transplantation is an effective option for metabolic correction of OTCD. Patients are able to discontinue dietary restriction, but previously existing neurodevelopmental problems do not improve.
Differential Diagnosis
It is imperative to distinguish OTCD from other genetic disorders that cause hyperammonemia. Hyperammonemia can arise from various conditions, including other UCDs, organic acidemias, defects in fatty oxidation, and disorders of pyruvate metabolism. Clinical indicators that narrow the differential to a UCD include respiratory alkalosis with a normal anion gap, normal blood sugar levels, and severely elevated ammonia levels.[27]
The differential diagnosis for a newborn male with hyperammonemia includes:
- N-acetyl glutamate synthase (NAGS) deficiency
- Carbamyl phosphate synthetase I (CPSI) deficiency
- Argininosuccinate synthetase (ASS) deficiency
- Argininosuccinate lyase (ASL) deficiency
- Fulminant hepatitis with fulminant liver failure [2]
The differential diagnosis for late-onset disease includes the following conditions (in addition to the ones listed above):
- Citrin deficiency
- Hyperornithinemia-hyperammonemia-homocitrullinuria syndrome
- Multiorgan failure due to hypoxic or ischemic injury
- Portal vein thrombosis
- Acute liver failure from other causes, such as acetaminophen toxicity [2]
Prognosis
OTCD causes high morbidity and mortality, particularly in individuals with neonatal onset.[28] According to a study, the mortality rate was 24% in those with neonatal onset of disease and 11% in those with a late onset.[9] The prognosis of a newborn in a hyperammonemic crisis depends on the duration of the elevated ammonia level.[2] A longer hyperammonemic state is associated with a worse prognosis. The peak ammonia level does not reliably predict prognosis, as those who are treated promptly with quick correction do better than those with a lower peak ammonia level and prolonged duration of hyperammonemia.[2] The presence or absence of seizure does not necessarily correlate with prognosis.[2] In contrast, blood ammonia and glutamine levels can be biomarkers for neurocognitive outcomes.[9]
Current European guidelines recommend a goal-of-care discussion in patients expected to have limited neurocognitive recovery before initiating treatment. Clinical parameters warranting this approach include hyperammonemic coma for 3 days or more, elevated intracranial pressure, and a peak ammonia level of 1000 µmol/L or greater.[6] As previously stated, the duration of hyperammonemia is a more reliable predictor of prognosis than the absolute value of peak ammonia levels.
Patients diagnosed early and treated emergently have improved prognoses, as do patients who adhere to low-protein diets and take medications that bypass the OTC enzyme in the urea cycle. The most common precipitant of clinical hyperammonemic crises is intercurrent infections.[9]
Patients with late-onset OTCD have better outcomes since they may have some functional OTC enzyme. Residual enzyme activity correlates with a later age of onset, lower initial peak plasma ammonium concentration, decreased frequency of hyperammonemic crises and predicts mortality.[29] The critical threshold of 4.3% residual enzymatic activity predicts mild/attenuated disease.[29]
Complications
Acute complications of OTCD include acute liver failure and severe hyperammonemic encephalopathy.[15] Patients may progress into a coma and require mechanical ventilation due to acute respiratory failure. Long-term complications include intellectual, neurological, and physical disabilities.[30] Case reports have documented hepatocellular adenoma in individuals with OTCD.[31]
Neuropsychological complications of OTCD include intellectual disability with developmental delay, attention-deficit/hyperactivity disorder (ADHD), as well as emotional and behavioral problems.[2] Studies have demonstrated that cognitive testing and intelligence scores correlate with peak ammonia levels and the frequency of hyperammonemic crises, underlining the impact of metabolic disturbances on cognitive outcomes.
These cognitive deficits can impact school performance, even when intellectual ability falls within the normal range.[2] Parents frequently report internalizing problems in about 50% of school-age children with this disease. These problems may manifest as withdrawal, depression, anxiety, or somatic complaints.[2]
Even heterozygous females, without a documented history of hyperammonemia, have been demonstrated to exhibit mild cognitive impairments, deficits in executive function, and challenges in fine motor tasks despite presenting with an average IQ on neuropsychological testing.[2] These findings underscore the importance of comprehensive neuropsychological assessments in individuals with OTCD, even in those considered carriers, to better understand the extent of cognitive and motor function implications.
A specific neurocognitive pattern of weakness in fine motor dexterity and speed, nonverbal intelligence, visual memory, attention, executive skills, and mathematics has been described in individuals with OTCD. The preservation of verbal intelligence, learning, and reading abilities accompanies this pattern.[6]
Deterrence and Patient Education
NBS for OTCD is not widely offered due to the limited feasibility attributed to the poor prognosis in early-onset cases, where symptoms often manifest before screening results are available.[6] Challenges such as the instability of glutamine in laboratory testing and the low specificity and sensitivity of low citrulline levels also contribute to the limitations of widespread screening. While reliable NBS is desirable for OTCD, current expert guidelines suggest insufficient data to recommend widespread NBS for OTCD.[6]
Upon identification of OTCD in a male or female proband, genetic counseling and screening of family members is indicated. This approach allows for identifying and treating asymptomatic individuals or those with mild symptoms, helping to prevent hyperammonemia.[2] Referral to a metabolic specialist, geneticist, and metabolic dietician is highly recommended.[6]
Nitrogen scavenger medication, urea cycle intermediate supplementation, and strict protein intake are necessary and recommended components of the management plan for these patients. While general guidelines provide protein intake thresholds based on age, determining the optimal protein threshold for each individual is variable and requires regular consultation with a metabolic dietician to prevent recurrent crises.[6]
Pearls and Other Issues
Key facts to keep in mind about OTCD are as follows:
- Early diagnosis and prompt intervention are crucial for favorable outcomes in OTCD.
- Genetic counseling and testing for family members help identify carriers and prevent potential complications.
- Delayed diagnosis may lead to severe complications and neurocognitive deficits.
- Newborn screening programs may aid in early detection and management.
- Inadequate adherence to dietary restrictions and medication regimens can result in metabolic decompensation.
- Collaborative care involving various specialists, such as geneticists, nephrologists, and dieticians, is essential for comprehensive management.
- Genetic counseling and testing for family members can inform carriers and guide preventive measures.
- Adherence to dietary restrictions, medications, and regular follow-ups is crucial for preventing hyperammonemic crises.
Enhancing Healthcare Team Outcomes
OTCD is a rare and potentially lethal condition. An informed and collaborative interprofessional team comprising clinical providers, nurses, pharmacists, and dieticians can dramatically impact patient outcomes. Given the genetic nature of this condition, aiding a patient also benefits others within their family. The patient’s care team members must collaborate efficiently in diagnosing and managing the patient, thereby preventing long-term neurocognitive deficits.
Symptoms may initially be discovered through the vigilance of neonatal nurses monitoring a patient’s vitals, parents observing their child, or individuals noticing unusual signs. In cases where patients present with neurological or behavioral disturbances, pediatricians, internists, and family physicians should promptly conduct ammonia levels testing.
Hemodialysis, crucial in emergent situations, underscores the importance of immediate nephrologist consultation. The involvement of geneticists, pharmacists, and dieticians is critical for short-term and long-term management. Given the genetic nature of the condition and its impact on family members, ethical considerations dictate recommending genetic counseling.
Timely intervention is key, as early detection allows for effective management, while delayed diagnosis may lead to fatal or debilitating outcomes. Collaborative efforts among physicians, nurses, dieticians, and pharmacists within an interprofessional team significantly improve clinical outcomes for individuals affected by this disease.