Continuing Education Activity
Optic nerve gliomas are benign tumors categorized as pilocytic astrocytomas and represent a significant portion of primary optic nerve tumors. They comprise around half of all primary optic nerve tumor cases and 1.5% to 4% of all orbital tumors. These tumors pose challenges in understanding their natural course and appropriate treatment strategies. They are part of a spectrum of primary tumors affecting the optic nerve, encompassing various entities such as optic nerve sheath meningioma and ganglioglioma. Optic pathway gliomas, including optic nerve gliomas, arise from the pre-cortical optic pathways and may present sporadically or in association with neurofibromatosis type 1 (NF1). Therefore, genetic testing for NF1 is recommended due to its association with optic pathway gliomas.
Although biopsy is usually unnecessary for diagnosis, advancements in molecularly targeted therapies may make genetic testing increasingly valuable. The initial evaluation includes magnetic resonance imaging of the brain and orbit to determine the tumor's location and guide treatment decisions. Management of optic gliomas necessitates an interprofessional healthcare team, including support from psychologists and ocularists, with evolving treatment options that emphasize achieving an optimal prognosis for affected patients. This activity reviews the evaluation of optive nerve gliomas in patients, with particular emphasis placed on the currently evolving treatment options and recommendations, highlighting the optimal prognosis for patients with this condition.
Objectives:
Identify the clinical features and imaging characteristics of optic nerve gliomas.
Implement evidence-based treatment strategies for optic nerve gliomas based on tumor location and patient characteristics.
Select appropriate surgical or medical interventions for optic nerve gliomas based on individual patient needs.
Collaborate with a multidisciplinary healthcare team, including neuro-ophthalmologists, oncologists, surgeons, and geneticists, to coordinate long-term follow-up and supportive care for patients with optic nerve gliomas and their families.
Introduction
Optic nerve gliomas are benign tumors categorized as pilocytic astrocytomas. They represent a significant portion of primary optic nerve tumors, comprising around half of all cases and 1.5% to 4% of all orbital tumors. Optic nerve gliomas and meningiomas continue to cause diagnostic controversy and pose challenges in understanding their natural course and appropriate treatment strategies, as their natural course is still being established. Treatments for both of these conditions continue to evolve.
The spectrum of primary tumors affecting the optic nerve is rare and encompasses various entities, including optic nerve glioma, malignant optic nerve glioma, optic nerve sheath meningioma, ganglioglioma, and primary lymphoma.
Etiology
Optic pathway gliomas are low-grade neoplasms arising from the pre-cortical optic pathways. These neoplasms can involve the optic nerve, optic chiasm, optic tracts, optic radiations, or the hypothalamus. They may present sporadically or in association with neurofibromatosis type 1 (NF1).[1] Therefore, genetic testing for NF1 is recommended due to its association with optic pathway gliomas. When associated with NF1, the inactivation of neurofibromin, a tumor suppressor on chromosome 17q, occurs, leading to the activation of the RAS signaling pathways and resulting in RAS-induced tumors.[2] Conversely, when arising sporadically, the most common genetic alteration identified is a BRAF-KIAA1549 fusion.[3]
Several studies have demonstrated that the prognosis of optic pathway gliomas in patients with NF1 is better with a better visual prognosis.[4] This subset of gliomas presents later and progresses over a longer course than spontaneous gliomas. Ophthalmic follow-up with vision assessment is necessary for all patients with gliomas, and electrophysiological assessment may be needed in very young patients where vision assessment can be challenging.
Epidemiology
Optic pathway gliomas primarily affect children aged 10 and younger, constituting 3% to 5% of childhood central nervous system (CNS) tumors.[2] However, these gliomas can occur across a wide age range, from birth to 79. Notably, 71% of cases occur within the first decade of life, and 90% are diagnosed within the first 2 decades. The mean age at which optic gliomas are diagnosed is approximately 8.8. In patients with NF1, approximately 15% to 20% eventually develop optic pathway gliomas, although only 30% to 50% of those individuals experience symptoms.[1][5] The incidence of NF1 in patients presenting with optic nerve gliomas varies widely, ranging from 10% to 70%, with an overall incidence of 29%.[6]
Men and women are generally affected equally by optic pathway gliomas.[7] However, with gliomas confined to the optic nerve, an increased incidence in women (65%) compared to men (35%) is common. Conversely, when tumors involve the optic chiasm, men and women are affected equally. At presentation, gliomas are confined to the optic nerve in 25% of cases, while 75% of cases show chiasm involvement.[8][9] Additionally, when the chiasm is involved, 40% of patients develop an extension of the tumor to the hypothalamus or third ventricle.
Histopathology
Gliomas were previously regarded as benign hamartomas with limited growth, but we now understand that their growth rate varies, and malignant transformations are rare occurrences. Additionally, these tumors tend to invade the leptomeninges, leading to their reclassification as true neoplasms capable of local invasion. Gliomas originate from astrocytes within the optic nerve and visual pathway. The proliferating astrocytes extend beyond the pia mater into the arachnoid and subarachnoid space, where fibrovascular and meningeal cell proliferation may occur—a phenomenon known as arachnoidal hyperplasia.
When exuberant, the appearance of tumor growth becomes visible. Tumor enlargement results from the proliferation of neoplastic cells due to reactive arachnoidal cell proliferation or the accumulation of mucosubstance. This mucosubstance is secreted by astrocytes and is positive for periodic acid–Shiff (PAS) staining. Additional histopathology results include:
- Usually, World Health Organization (WHO) grade I pilocytic astrocytoma with immature astrocytes
- Tumor cells exhibiting spindle-shaped nuclei and a pilocytic or hair-like appearance
- Biphasic pattern with varying proportions of piloid areas alternating with spongy areas
- Intracytoplasmic Rosenthal fibers and eosinophilic granular bodies
- Microcystic areas
- Focal calcification may be seen
- Rosenthal fibers characterized by tapered corkscrew-shaped, brightly eosinophilic, hyaline masses [10]
These tumors are typically diagnosed as anaplastic astrocytomas (WHO grade III) or glioblastomas (WHO grade IV). Histologically, they exhibit malignant astrocytes with pleomorphic nuclei, necrosis, and vascular proliferation, often accompanied by invasion into the surrounding brain tissue.
History and Physical
The location of the tumor determines the presenting signs and symptoms.
Loss of Vision
Children often do not complain of loss of vision. Approximately 85% of patients with glioma will experience some degree of vision loss; over time, around 25% will retain vision between 20/20 and 20/40. About 60% of patients will develop vision worse than 20/300. Patients may also exhibit an afferent pupillary defect. Where visual field assessment is possible, visual field defects may be observed. Optic nerve atrophy (seen in 60% of patients upon fundoscopic examination) and optic disc edema (seen in 50% of intraorbital tumors) may be present. Patients with chiasmal tumors will exhibit optic nerve edema in 20% of cases, especially those with associated orbital optic nerve involvement.
Proptosis
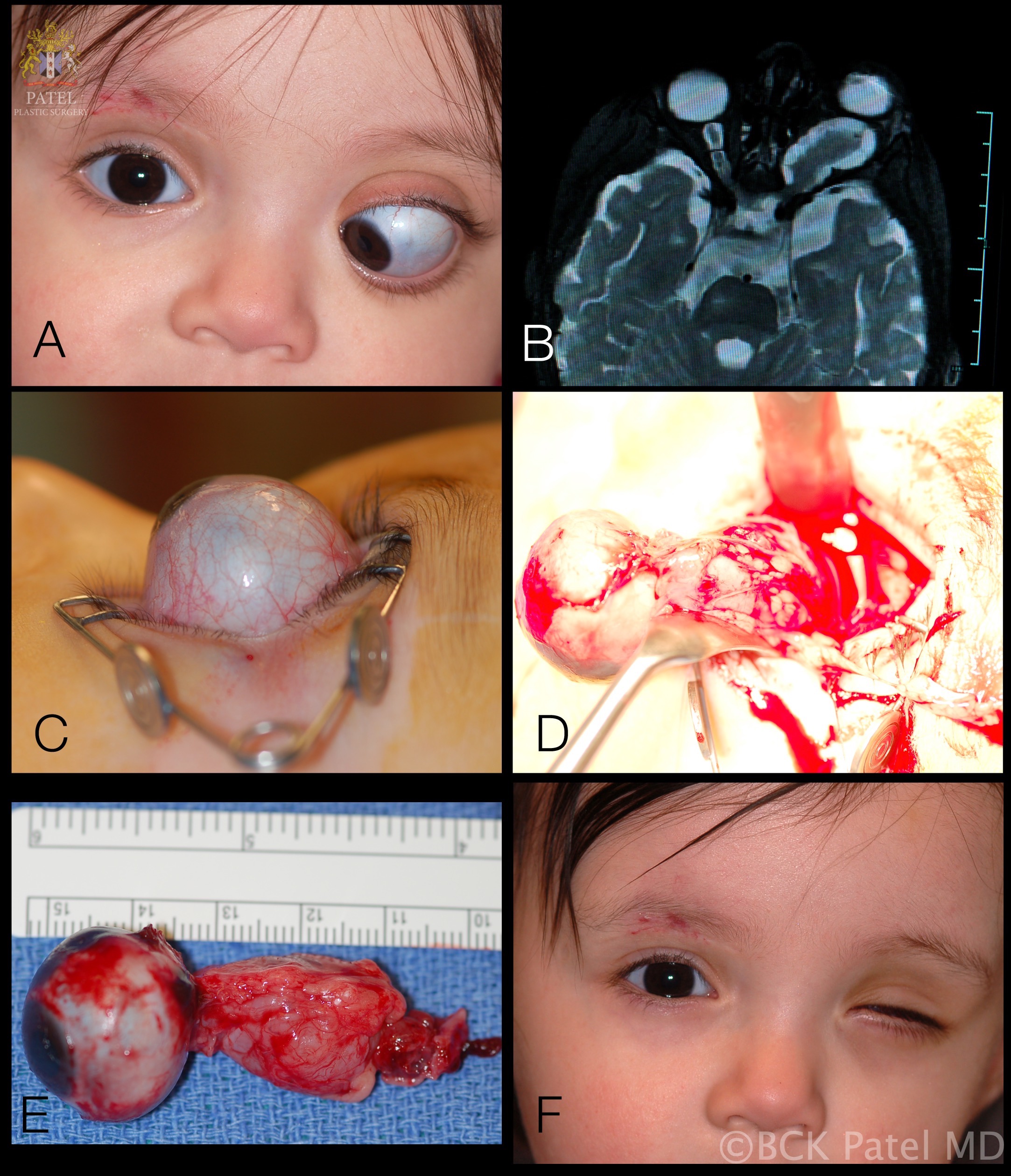
Proptosis is the most common presenting sign in 95% of patients with optic glioma. However, it is less commonly observed when the tumor is confined to the optic chiasm (seen in less than 20% of patients). In cases where proptosis is seen in patients with optic chiasm glioma, the optic nerve has almost always concomitant involvement. Ocular motility issues with optic nerve gliomas are infrequent. Limitation of ocular motility may be observed in 30% of optic nerve gliomas confined to the orbit and in 20% of cases where the chiasm is involved.
Headache/Pain
The most common presenting symptom of optic pathway gliomas is headache, although it is only found in about 30% of patients, predominantly in cases involving the chiasm. Other symptoms and signs include nystagmus, spasmus nutans, convulsions, nausea, dizziness, strabismus, developmental regression, and growth retardation. Hydrocephalus may develop as the tumor spreads from the chiasm. Additionally, these patients may experience hypothalamic-pituitary dysfunction, leading to precocious puberty, growth hormone deficiency, and deficiencies of gonadotropin, thyroid-stimulating hormone (TSH), and adrenocorticotropic hormone (ACTH).
In summary, the optic pathway gliomas present with:
- Early vision loss (88%)
- Optic disc swelling (35%)
- Optic nerve head atrophy (59%)
- Proptosis in orbital tumors (94%)
- Proptosis in chiasmal tumors (22%)
- Nystagmus (24%)
- Hypothalamic signs (26%)
- Increased intracranial pressure (27%)
Evaluation
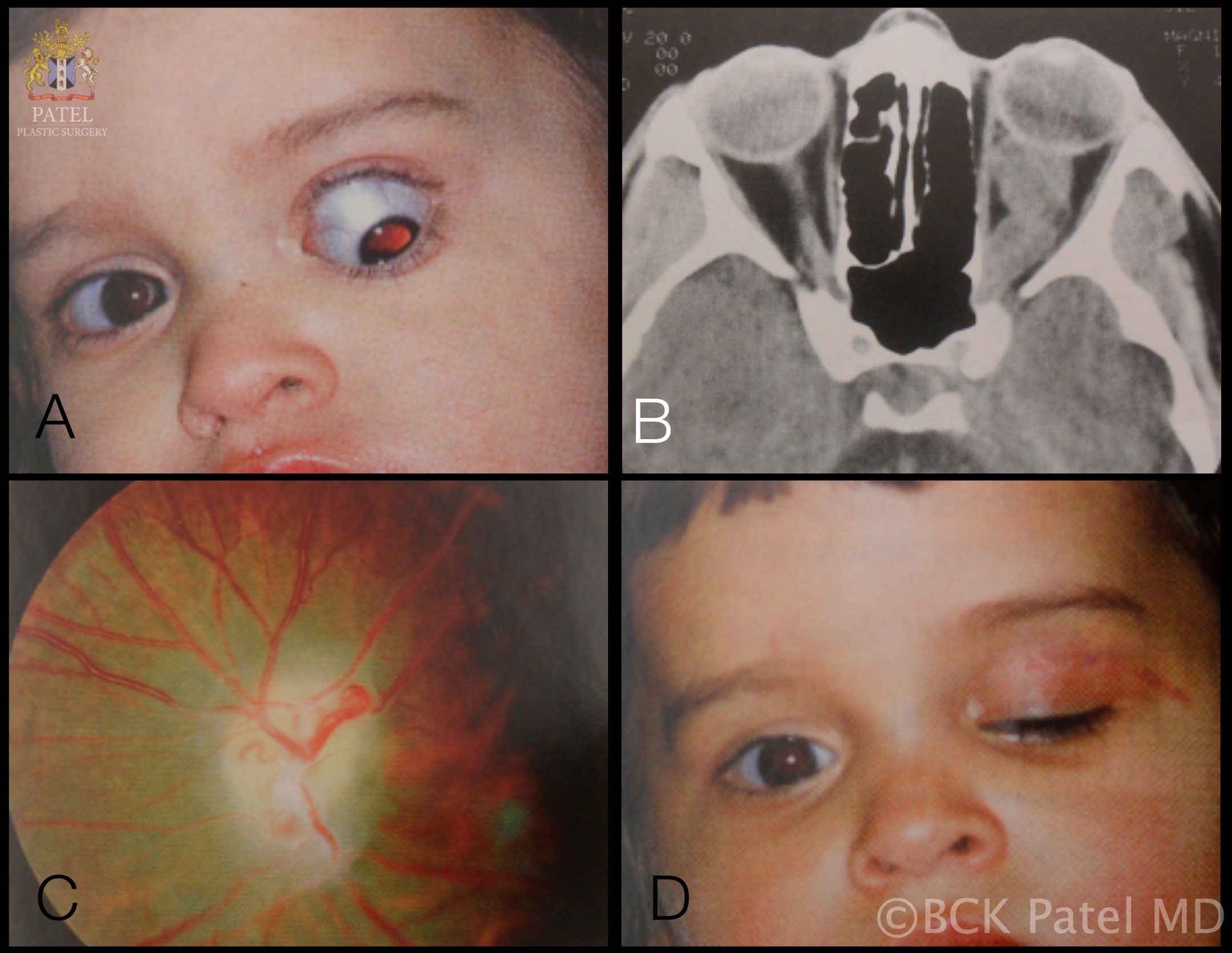
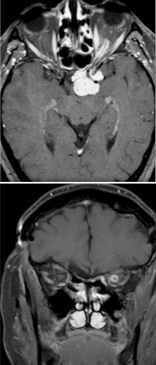
The initial workup typically includes magnetic resonance imaging (MRI) of the brain and orbit. Robert-Boire et al found that approximately 80% of patients exhibit radiographical progression on MRI.[1] Enlargement of the optic canal is observed in 80% of patients with optic nerve involvement. In 25% of patients with chiasmal glioma, enlargement of the sella turcica and J-shaped excavation may be evident (see Image. Optic Nerve Glioma in a Boy). When evaluating chiasmal, hypothalamic, and optic tract tumors, MRI is superior to computed tomography (CT).
On MRI, gliomas typically exhibit slightly prolonged T1 relaxation times, with the tumor image appearing isointense or slightly hypointense compared to the normal optic nerve. T2-weighted images show a hyperintense image with prolonged T2 relaxation time. CT scans reveal an enlarged optic nerve and/or chiasm. Contrast-enhanced imaging shows mild-to-moderate enhancement, which is less pronounced than that seen with optic nerve sheath meningiomas. The optic nerve typically demonstrates fusiform swelling, although rounded and exophytic changes may also be visualized. Calcification in optic pathway gliomas is uncommon but may be observed (see Image. Left Optic Nerve Glioma in a Toddler).
Genetic testing for neurofibromatosis is always warranted. A biopsy is typically unnecessary because the diagnosis relies on imaging and clinical examination. However, new molecularly targeted therapies may increase the utility of genetic testing for the neoplasm. Both optic nerve gliomas and meningiomas can cause diffuse enlargement of the optic nerve, as well as a globular or fusiform enlargement. However, meningiomas exhibit "tram-tracks," resulting from thickened and denser optic nerve sheath, creating central lucency representing the residual optic nerve (see Image. Optic Nerve Sheath Meningioma). Although calcification is observed in 20% to 50% of meningiomas, it is rare in gliomas. Meningiomas demonstrate increased signal intensity compared to the normal nerve on both T1- and T2-weighted MRI sequences. In contrast, optic nerve gliomas appear isointense or slightly hypointense on T1-weighted sequences and hyperintense on T2-weighted sequences.
Treatment / Management
Understanding the natural history and course of these gliomas is crucial for appropriate management. Gliomas exhibit slow growth but can potentially spread into surrounding tissues, including around the optic nerve, the chiasm, and the adjacent brain. Vision stabilizes in most patients with slow growth. However, about 40% of patients may experience continued tumor growth. Diagnosis is only achievable through a biopsy and subsequent histopathological examination.
Depending on the case, treatment is overseen by an interdisciplinary healthcare team consisting of neuro-ophthalmologists, hematology-oncologists, oculoplastic surgeons, and radiation oncologists. Management strategies are tailored based on several factors, including symptoms, tumor characteristics, progression, changes in vision, clinical course, NF1 status, neoplasm genetics, patient age, and patient preference. Observation is typically the initial approach until significant visual deficits or radiographical progression occurs.
In 80% of patients, an initial phase of visual deterioration is followed by stabilization. However, stable vision does not always equate with a stable tumor, as tumor progression can occur in these patients. Conservative follow-up without intervention has a good long-term survival prognosis.[11][12] Typically, young patients and those with NF1 undergo a period of observation, with tumor size monitored using regular interval MRIs and regular vision assessments. Any change in clinical status or imaging findings would prompt the initiation of treatment.
Management Approach for Gliomas
Optic nerve glioma
- Clinicians typically opt for clinical and radiological observation if the tumor is stable.
- Surgical intervention is considered in cases of vision loss and severe proptosis.
- Surgical resection may be considered if there is a posterior progression of the tumor with a risk of invasion of the chiasm or surrounding tissues.
Optic chiasm glioma
- If the glioma is stable, healthcare providers recommend observation with sequential radiological assessment.
- If progression occurs in patients aged 10 or younger, healthcare providers may consider chemotherapy.
- If progression occurs in patients aged 10 or older, healthcare providers may recommend a combination of chemotherapy and radiotherapy.
Midbrain glioma
- If the glioma remains stable in patients for more than 10 years, healthcare providers consider chemotherapy and radiotherapy.
- If the glioma remains stable in patients aged 10 or younger, healthcare providers consider chemotherapy.
- In case of progression, healthcare providers consider a combination of chemotherapy and radiotherapy.
Malignant optic nerve glioma
A rarer type of glioma is seen in older patients. Since Hoyt et al first described aggressive optic nerve glioma in adults, typically malignant, an additional 60 cases have been reported.[13] The age range for these tumors is 6 to 79, with the majority of occurrences happening in middle age, characterized by a mean age of presentation at 48.[14] Males are affected in 65% of cases. The primary site of tumor occurrence is the optic chiasm, which is nearly always bilateral. Extension of the malignant tumor from the chiasm into the orbital optic nerve is observed in 23% of cases. In 50% of cases, the tumor extends posteriorly from the chiasm, along the optic tracts, into the hypothalamus or the temporal lobe.
Clinical Course
Patients with malignant optic nerve gliomas typically present with rapid vision loss, initially unilaterally and then bilaterally. Upon initial evaluation, 63% of cases demonstrate vision deterioration in both eyes. Due to this presentation, the condition may be misdiagnosed as anterior ischemic optic neuropathy or optic neuritis. Most patients exhibit optic disc edema, which may progress to optic atrophy if the patient survives. Unlike optic pathway gliomas, proptosis is observed in only 20% of patients with malignant optic nerve gliomas. In 80% of cases, CT or MRI imaging will reveal optic chiasm enlargement and enhancement, along with the involvement of intracranial portions of the nerves. Diagnosis is only achievable through a biopsy and subsequent histopathological examination.
Currently, neither surgery nor radiotherapy significantly alters the prognosis of this condition. However, in rare cases, aggressive treatment combining radiotherapy, chemotherapy, and surgery may marginally improve survival by a few months.
Differential Diagnosis
Optic nerve gliomas can typically be diagnosed accurately using MRI due to their unique features. Optic nerve sheath meningiomas may share a similar location, but they usually exhibit distinct radiographical features and are rare in children. The differential diagnosis should, therefore, include conditions such as meningioma, rhabdomyosarcoma, orbital lymphoma, benign lymphoproliferative lesions, neuroblastoma, epithelial tumors, inflammatory lesions, infectious lesions, and metastasis of extraocular tumors.
Surgical Oncology
Surgery
After the mainstay of treatment, the role of surgery is largely confined to patients with progressive vision loss, painful keratopathy, severe proptosis, worsening hydrocephalus, or in cases where the tumor extends posteriorly, posing a threat to the optic chiasm. Optic nerve gliomas rarely progress to involve the chiasm or affect contralateral fibers.[15] Surgical resection is beneficial after vision loss to reduce proptosis and alleviate orbital pain. The tumors are typically excised using the lateral trans-orbital approach to preserve the globe and all extraocular muscles whenever feasible for optimal cosmetic outcomes. However, if surgery is conducted while vision is preserved, there is a substantial risk of vision loss.
Outcomes of Resection by Subtype
Gliomas confined to the optic nerve: When these tumors, which are confined to the optic nerve, are treated conservatively or incompletely resected, 17% of patients experience recurrence or progression. Among this group, 12% experience mortality due to intracranial extension. Overall, the prognosis for vision is favorable, with more than 90% of patients maintaining stable vision over time.
Surgical resection of optic nerve gliomas, with or without radiotherapy, significantly reduces the mortality rate to nearly zero. However, it is noteworthy that there is a risk of vision loss associated with surgery. Resection of optic nerve gliomas with posterior extension and vision loss helps prevent further tumor progression and reduces mortality rates.
Gliomas extending to the chiasm: If invasion of the adjacent third ventricle or hypothalamus is absent, these tumors exhibit behavior similar to those confined to the orbital optic nerve. Chiasmal gliomas have a mortality rate of 17% over a period of 10 years if not treated. Mortality rates increase if the tumor extends into surrounding tissues such as the hypothalamus or third ventricle.
Partial resection of the tumor from the chiasm may result in a high recurrence or progression rate (64%). Despite this, the visual prognosis is generally favorable, with 80% of cases remaining stable without treatment. Fractionated radiation therapy can help slow tumor growth in the chiasm without the risk of vision loss associated with surgery. Overall, chiasmal gliomas have a mortality rate of 22%, a recurrence or progression rate of 43%, and visual stabilization is achieved in 68% of cases when considering all treatment modalities.
Gliomas extending to the chiasm and invading the hypothalamus: The mortality rate in patients with chiasmal tumors extending into the hypothalamus or third ventricle exceeds 50% over a 15-year period. Although mortality may be reduced with radiotherapy and chemotherapy, recurrence or progression still occurs in 52% of cases.[6]
Complications
The overall surgical mortality is approximately 6%.[16] Surgical complications include vision deterioration, multiple endocrine deficiencies, and hypothalamic dysfunction.[17]
Radiation Oncology
Radiation
Considering that most optic pathway glioma patients are aged 20 or younger, offering radiotherapy necessitates careful consideration. Balancing the potential benefits of local control with long-term toxicity is crucial in treatment decision-making for this age group. Typically, radiotherapy is indicated for patients who have experienced disease progression despite systemic chemotherapy, children aged 10 or younger, cases with intracranial extension, progressive disease at initial diagnosis in older patients, or postoperative treatment. Extensive research has shown acceptable overall survival and progression-free survival rates with prolonged systemic chemotherapy, relegating radiotherapy primarily to salvage treatment.[18][19]
Dosing
Typically, the dosing ranges between 45 and 54 Gy in fractions of 1.8 Gy each, with adjustments made based on the ability to meet dose constraints for nearby organs at risk.
Simulation
Simulating and treating pediatric patients can be challenging, possibly requiring anesthesia for very young children. The radiotherapy vault can be intimidating, so efforts should focus on creating a child-friendly treatment area. Immobilization is performed using a custom thermoplastic head mask, which may cause distress in younger patients and needs careful management.
Target Delineation
MRI imaging and fusion with CT simulation scans are crucial for target delineation, especially utilizing T1 post-contrast and T2-FLAIR sequences. The gross tumor volume (GTV) is defined by abnormalities seen in these sequences. Optic pathway gliomas often exhibit variable enhancement on T1-contrast and central hyperintensity on T2 sequences. A 10-mm margin was traditionally added to the GTV to form the clinical target volume. However, recent pediatric protocols such as ACNS 0221 aim to reduce this expansion to 5 mm to minimize normal tissue radiation and late toxicity. Additionally, a planning target volume (PTV) expansion of 3 to 5 mm is included.[20]
Dose Constraints
For the target PTV, the goal is to cover 100% of the PTV volume with at least 95% of the prescription dose and less than 10% of the volume receiving more than 107%. QUANTEC dose constraints offer dose-dependent risk estimations for various normal tissues, particularly those in the brain.[21] Organs should be contoured, including the optic nerve, chiasm, retina, brainstem, and uninvolved brain.
Proton Therapy
Proton therapy has garnered significant interest, especially in treating pediatric tumors. The unique physical properties of protons compared to x-ray photons, especially the manner of dose deposition in the form of a Bragg peak and the lack of an exit dose, may reduce the volume of normal tissue irradiated and, thus, late toxicity. Dosimetric studies comparing 3D conformal photons with protons showed a 47% decrease in optic nerve dose, an 11% decrease in chiasm dose, a 13% decrease in pituitary gland doses, and a 39% decrease in temporal lobe dose compared to photon plans.[22]
Oncologic Outcomes
Patients treated with radiotherapy have a 5-year progression-free survival ranging from 48% to 100% and a 5-year overall survival ranging from 79% to 96%.[2] Overall survival rates remain similar regardless of radiotherapy administration. Radiographic responses are not immediate, typically taking about 5 years to observe. Factors impacting progression-free survival include tumor extension to the optic chiasm and age 15 or younger.[23] Treatment results in stabilized or improved vision in 81% of patients. A retrospective analysis of pediatric low-grade gliomas (including 52% optic pathway gliomas) treated with proton therapy displayed a 5-year progression-free survival of 84% and an overall survival of 92%, comparable to most photon therapy outcomes.[24]
Late Toxicity
Due to their occurrence in younger patients, concerns about late toxicity from radiation treatment for optic pathway gliomas are significant, as these effects may take years to manifest. Consequently, radiotherapy is often reserved for salvage treatment due to concerns about potential long-term effects.
Secondary Malignancies
The development of a secondary CNS tumor usually occurs within the high-dose radiation field with a mean time to development of 6 to 9 years and a 10-year cumulative incidence of 8%.[25] The risk is particularly high in patients with NF1, with 50% of the irradiated patients developing a second CNS tumor.[26] These secondary tumors can include anaplastic astrocytoma and glioblastoma multiforme.[27] Proton beam therapy shows promise in reducing the incidence of secondary malignancies compared to photon-based treatments. A study of 1713 children treated with proton beam therapy reported a 10-year cumulative incidence of 3.1%. Additionally, an analysis of the SEER database suggested a lower incidence of second malignancies in the proton therapy group compared to the photon therapy group (5.2% versus 7.5%).[28]
Intellectual Impairment
Cranial radiotherapy has been linked to adverse effects on intellectual abilities, especially when administered at a younger age. Studies indicate that approximately 30% of patients treated with cranial radiation for chiasmatic gliomas exhibit intellectual deficits.[29] Neuropsychological assessments of children with CNS malignancies who underwent radiation therapy revealed lower overall intellectual functioning compared to those without radiotherapy. Among the affected areas, non-verbal abilities showed significant impairment.[30] Apart from age at treatment initiation, radiation doses to the hippocampus and temporal lobe played roles in neurocognitive decline.[31] Proton beam therapy has shown promise in mitigating cognitive decline by reducing radiation exposure to these critical brain areas.[32]
Visual Deficiency
Given the location of these tumors, avoiding exposure to the uninvolved optic nerve is challenging after full-dose radiation treatment. According to QUANTEC, the risk of optic neuropathy is <3% with doses <55 Gy, although hotspots in the optic pathway can surpass this limit despite a maximum dose of 54 Gy for optic pathway gliomas. Risks escalate from 3% to 7% and 7% to 20% for doses of 55 to 60 Gy and >60 Gy, respectively.[33] Retrospective analyses estimate long-term visual decline risks at 7% to 17%. However, delays in radiotherapy may compromise vision preservation.[34] Proton therapy shows promise in significantly reducing blindness rates, which is evident in a study reporting a 100% 8-year blindness-free survival rate.[35]
Hypothalamic-Pituitary Insufficiency
One of the most common late complications post-radiotherapy is endocrine derangement. In total, 72% of patients treated with cranial radiation develop central endocrine insufficiency, with growth hormone and TSH deficiency being the most prevalent. Approximately 20% of patients experience ACTH deficiency, delayed puberty, or panhypopituitarism.[36] Obesity and diabetes insipidus are also observed. Early identification and treatment with exogenous hormone supplementation can prevent more permanent developmental complications.
Vasculopathy
Approximately 18% of radiated patients experience occlusive vasculopathy, contrasting with no cases in the unirradiated group.[37][38] Angiographically, the condition resembles Moyamoya disease with a median onset of 3 years. A reported dose-response relationship of a 5% hazard per Gray of radiation exists.[39] Due to tumor geometry, avoiding major cerebral vasculature, such as the Circle of Willis, is challenging. Ischemic strokes may occur, particularly in chiasmal tumor treatments. Despite advances in radiation techniques, this risk remains relatively constant. Proton beam therapy may slightly reduce incidence to 5% to 10%.[40]
Medical Oncology
Systemic Chemotherapy
Systemic chemotherapy becomes the primary treatment for optic pathway gliomas, particularly in younger patients, when evidence of progressive disease and/or vision compromise is evident. This approach takes precedence due to the late toxicities associated with radiotherapy, potential surgical complications, and favorable response rates to systemic therapies. Prospective trials have explored a chemo-first strategy with alternating regimens, including procarbazine/carboplatin, etoposide/cisplatin, and vincristine/cyclophosphamide, administered every 3 weeks. The 5-year progression-free survival rate was 34%, with an overall survival rate of 89%. In total, 39% of patients required salvage radiotherapy at 5 years.[19] In most cases, chemotherapy with vincristine/carboplatin is considered first-line treatment with a 3-year progression-free survival of 68%, and tumor response appeared to depend upon age. Patients aged 5 and older have a 39% progression-free survival benefit at 5 years.[41] Additional chemotherapy regimens, including thioguanine/procarbazine/CCNU/vincristine (TPCV), cisplatin/etoposide, and temozolomide, are also utilized. Randomized comparisons of TPCV versus vincristine/carboplatin showed higher rates of 5-year event-free survival, although statistical significance was not reached.[42]
Monotherapy with vinorelbine has shown promise in managing progressive optic pathway gliomas, achieving a 5-year progression-free survival rate of 34% to 42%. Similarly, vinblastine monotherapy has demonstrated a 5-year progression-free survival of 53%, although it encompasses various tumor histologies. However, both therapies carry risks of hypersensitivity reactions, gastrointestinal disturbances, ototoxicity, nephrotoxicity, neurotoxicity, myelosuppression (platinum-based chemotherapy), and a heightened risk of secondary leukemia, notably with CCNU and procarbazine. Vincristine use may lead to peripheral neuropathies, hyponatremia, and alopecia. While TPCV is generally avoided in NF1 patients, it remains an option for spontaneous optic pathway glioma treatment.
Targeted Therapies
Optic pathway gliomas are often quite vascular, and vascular endothelial growth factor (VEGF) promotes vessel formation. Targeted therapies such as bevacizumab, a VEGF inhibitor, have shown efficacy in controlling the growth of optic pathway gliomas due to their vascularity. Bevacizumab can be used alone or in combination with vinblastine or irinotecan. Bevacizumab monotherapy has yielded positive outcomes in 86% of patients, improving visual symptoms and showing objective responses. Some data suggest monotherapy may have similar efficacy to combination therapy with less toxicity. Combination therapy with irinotecan demonstrated a 2-year progression-free survival of 47.8%. Toxicities related to bevacizumab include hypertension, fatigue, proteinuria, and bleeding.
Targeted therapies focusing on the mitogen-activated protein kinase (MAPK) pathway have become more prevalent, emphasizing the importance of tumor biopsies. The most common genetic aberration is a BRAF-KIAA1549 proto-oncogene fusion, which activates the MEK/MAPK/ERK and mTOR pathways and causes cell proliferation, survival, and tumorigenesis. MEK inhibitors such as selumetinib, trametinib, and refametinib target this pathway and report a 69% 2-year progression-free survival for refractory optic pathway gliomas.[43] Trametinib is currently the most widely used MEK inhibitor, although more than a dozen alternatives are undergoing clinical trials.[44] These agents represent a viable salvage alternative for optic pathway gliomas that have progressed through other systemic agents. The most common adverse effects of MEK inhibitors involve dermatologic conditions; however, vision-threatening ocular adverse events, including optic neuropathy, retinal vein occlusion, uveitis, neurosensory retinal detachment, and MEK inhibitor-associated retinopathy, have been reported. Fortunately, these events are rare and often reversible, particularly in pediatric patients.[15]
Prognosis
NF1-associated optic pathway glioma generally carries a better prognosis, although this outlook is influenced by patient selection bias and tumor location. Many optic pathway gliomas exhibit minimal or no progression over time and may remain asymptomatic throughout a patient’s life. Such tumors are often incidentally discovered in NF1 patients but tend to be symptomatic in sporadic cases.[45] NF1-associated gliomas are more frequently located pre-chiasmatically. Research by Robert-Boire et al indicates that progression rates are comparable between NF1 and sporadic cases. However, among 40 patients with optic pathway gliomas, those with bilateral chiasmatic or post-chiasmatic sporadic tumors were the only ones to experience progression-related mortality.[1]
A comprehensive literature review conducted by Jonathan Dutton has revealed that across all patients with optic pathway gliomas and various treatment modalities (including observation), tumor recurrence or progression occurs in 38% of cases.[6] Tumor-related mortality with a mean follow-up of 11 years is 36%. Approximately 55% of patients either maintain stable vision or show improvement, while 45% experience progressive vision loss. Spontaneous regression of optic gliomas is infrequent but has been documented in some cases.[46] Malignant transformation of optic pathway gliomas, although exceedingly rare, has also been reported in the medical literature.[47]
Unfortunately, all patients with malignant optic nerve gliomas progress to complete vision loss within months of presentation. The mortality rate is 100%, with a mean survival period of less than 1 year.
Complications
Surgical complications include vision deterioration, multiple endocrine deficiencies, and hypothalamic dysfunction.[17] Monotherapy with vinorelbine or vinblastine carries the risk of hypersensitivity reactions, gastrointestinal disturbances, ototoxicity, nephrotoxicity, neurotoxicity, myelosuppression (platinum-based chemotherapy), and a heightened risk of secondary leukemia, notably with CCNU and procarbazine. Vincristine use may lead to peripheral neuropathies, hyponatremia, and alopecia. Toxicity to therapy with bevacizumab can result in hypertension, fatigue, proteinuria, and bleeding. The most common adverse effects of MEK inhibitors involve dermatologic conditions; however, vision-threatening ocular adverse events, including optic neuropathy, retinal vein occlusion, uveitis, neurosensory retinal detachment, and MEK inhibitor-associated retinopathy, have been reported. Fortunately, these events are rare and often reversible, particularly in pediatric patients.[15]
Post-radiotherapy may cause endocrine derangement. In total, 72% of patients treated with cranial radiation develop central endocrine insufficiency, with growth hormone and TSH deficiency being the most prevalent. Approximately 20% of patients experience ACTH deficiency, delayed puberty, or panhypopituitarism.[36] Obesity and diabetes insipidus are also observed. Early identification and treatment with exogenous hormone supplementation can prevent more permanent developmental complications.
Enhancing Healthcare Team Outcomes
Management of gliomas necessitates an interprofessional healthcare team consisting of neuro-ophthalmologists, hematology-oncologists, orbital and oculoplastic surgeons, radiation oncologists, and specialty-trained nurses. An experienced electrophysiologist may also be required in cases where vision assessment may be challenging, especially in young children. In addition, clinical psychologists play a crucial role in clinic visits and during surgical or treatment procedures. Early introduction of the family to an ocularist is beneficial, especially before any potential globe-removal surgery. Education about monocular vision is essential when dealing with unilateral vision loss.