Continuing Education Activity
Maple Syrup Urine Disease (MSUD) is a rare genetic disorder impacting branched-chain amino acid metabolism. This mutation is tested for perinatally, but a high suspicion must be present given this entity's significant and potentially irreversible developmental effects. Causes of mortality include cerebral edema, seizures, coma, respiratory failure, and metabolic derangements. Early recognition and treatment are crucial to reducing the morbidity and mortality of this disease. Designed for healthcare professionals, this course covers MSUD's pathophysiology, clinical manifestations, diagnosis, and management strategies. Healthcare professionals will gain insights into early recognition, dietary therapy, acute metabolic decompensation management, and long-term care, enhancing their ability to provide optimal care for patients with MSUD. Through this activity, clinicians will be better equipped to coordinate care with interprofessional team members and contribute to improved patient outcomes and quality of life.
Objectives:
Identify the early clinical manifestations of maple syrup urine disease to facilitate prompt diagnosis.
Implement a well-structured management plan for patients with maple syrup urine disease.
Evaluate patients for complications of maple syrup urine disease.
Communicate with interprofessional healthcare team members to care for patients with maple syrup urine disease.
Introduction
Maple syrup urine disease (MSUD) is a defect of amino acid metabolism due to abnormal activity of the branched-chain alpha-ketoacid dehydrogenase (BCKAD) complex. This complex is responsible for breaking down the 3 essential branched-chain amino acids (BCAA): isoleucine, leucine, and valine. These BCAA are usually used for energy production or increased protein (ie, muscle) synthesis through their effect on the mTOR signaling pathway.[1] In the brain, BCAA's function is to promote protein and neurotransmitter synthesis and energy production.[2]
MSUD, also known as branched-chain α-ketoacid dehydrogenase deficiency, occurs when an underlying defect in the BCKAD complex located in the cellular mitochondria disrupts the metabolism of BCAA, which leads to their accumulation in the plasma, brain, and other tissues as well as their respective derivative ketoacids in the urine.[3] It classically manifests in the neonatal period with failure to thrive, delayed developmental milestones, feeding difficulties, lethargy, irritability, and a maple syrup odor first noticeable in the cerumen and then the urine.[3] If left untreated, the most severe, classic form of MSUD can quickly lead to irreversible neurological injury manifesting as brain damage, seizures, a coma, or central respiratory failure within just 7 to 10 days after birth. Death in untreated individuals with the severe form of the disorder occurs within 2 months. Good clinical outcomes can be expected if management is initiated early and expectantly, but any significant lapse in treatment can result in permanent brain damage or worse. Long-term treatment consists of significant, lifelong dietary restrictions of branched-chain amino acids and close, continuing metabolic monitoring, but acute exacerbations with decompensation may occur, requiring emergency management.
Etiology
MSUD is caused by an autosomal recessive mutation in 1 of the 3 genes: BCKDHA, BCKDHB, or DBT. Branched-chain ketoacid dehydrogenase (BCKAD) is located within the inner mitochondrial membrane of various tissues such as skeletal muscle, liver, kidney, and the brain. It comprises 3 catalytic subunits (E1, E2, and E3). BCAA transaminase helps mediate the catabolism of BCAA. Most of this activity occurs in the mitochondria where BCKAD is located. One metabolic product of leucine is alpha-ketoisocaproic acid, which acts as a neurotoxin and contributes to the encephalopathic syndrome.[3][4]
Epidemiology
MSUD has an estimated worldwide incidence of just 1 case per 185,000 live births.[5] For perspective, the Health Resources and Services Administration estimates only 30 new neonatal cases a year are diagnosed in the United States. Higher occurrences have been noted in populations with a higher rate of consanguinity. MSUD affects males and females equally. In the Ashkenazi Jewish population, the incidence is estimated at 1 in 26,000 live births. In Mennonites, MSUD occurs with an incidence of 1 in 380 newborns. This is often termed a "founder effect" in the BCKDHA (E1a) gene. In the Portuguese Romani population, incidence is estimated at 1 case per 71 births.[6]
Pathophysiology
MSUD occurs due to a pathogenic defect in any BCKAD subunit, resulting in elevated branched-chain amino acids and their corresponding alpha keto-acids. Accumulated BCAA and alpha-ketoacids manifest as a constellation of clinical symptoms due to the central nervous system, immune system, and skeletal muscle dysfunction. The BCAA (ie, isoleucine, leucine, and valine) are essential amino acids with hydrophobic side chains in protein-rich food. The catabolism of these amino acids is necessary to maintain various physiologic functions, including:
- Cellular signaling
- Cholesterol synthesis
- Energy production
- Fatty acid synthesis
- Gluconeogenesis
- mTOR pathway regulation
- Protein synthesis
The liver and kidney are responsible for the catabolism of 10% to 15% of BCAA.[7] Most BCAA transamination and oxidation occur in skeletal muscle.[8] Leucine comprises about 11% of all human tissue protein, about 110 g/kg of body weight. During periods of catabolism, serum levels can increase very rapidly. Sotolone, an isoleucine metabolite, is responsible for the maple syrup odor of the cerumen and urine.[9] Elevated leucine and alpha-ketoisocaproic acid levels notoriously cause neurochemical disturbances, resulting in clinically apparent neurotoxicity. Neurotoxicity is aggravated by the metabolic decompensation in MSUD, which activates matrix metalloproteinases, resulting in the further breakdown and dysfunction of the blood-brain barrier.[4][10]
High intracranial leucine concentrations compete with the cerebral uptake of several other important amino acids, particularly phenylalanine, glutamine, histidine, methionine, and tryptophan, negatively impacting brain growth, neurotransmitter production, and myelin synthesis.[11][12][13] The restricted supply of essential amino acids leads to decreased neurotransmitters, including dopamine, serotonin, norepinephrine, epinephrine, GABA, and glutamate. Alpha-ketoisocaproic acid levels greater than 60 mmol/L negatively affect astrocyte transamination reactions.[13][1][13] This restriction causes low cerebral glutamate levels, which results in cognitive dysfunctions (eg, learning disabilities and memory loss). Furthermore, elevated leucine concentrations impair cell volume regulation. This results in decreased blood osmolarity, low sodium concentrations, and increased intracellular water, leading to cerebral edema.[14]
In infants and children, decreased blood osmolarity can exacerbate cerebral edema and further precipitate brain herniation. Experts theorize that the citric acid cycle in the brain is destroyed by BCAA inhibition of pyruvate and α-ketoglutarate dehydrogenase, as well as the mitochondrial respiration chain, which affects amino acid and protein synthesis, causing abnormal myelination and cerebral edema.[15] Long-term exposure to high levels of BCAA will cause myelin dysplasia.[16][17]
History and Physical
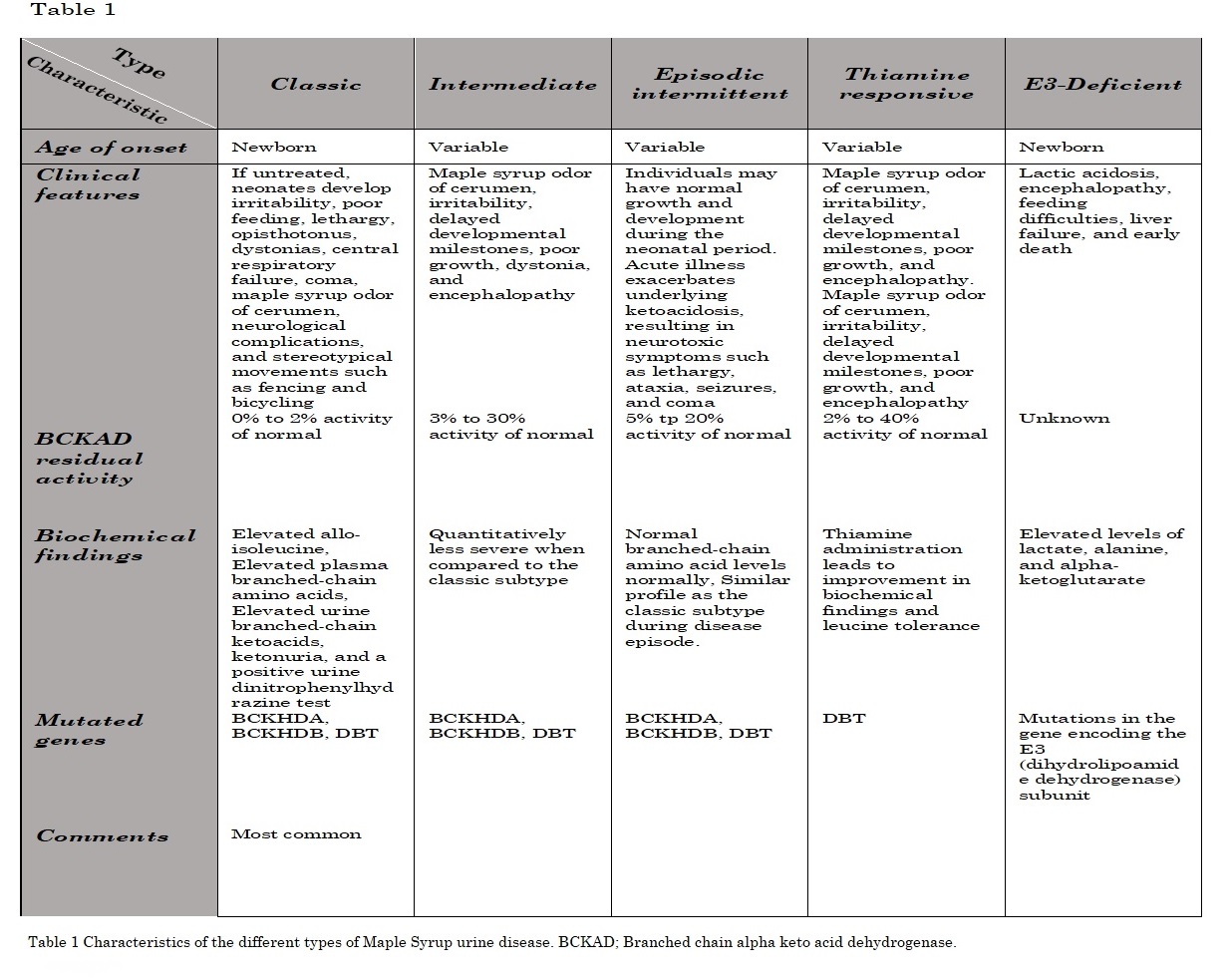
Patients are clinically distinguished based on their clinical presentation, age of onset, and residual BCKAD enzyme activity.[5] There is no good correlation between genotype and phenotype. The clinical presentation relies on the BCKAD residual function.[3] However, phenotypic classification depends on the response to metabolic decompensation and leucine tolerance (see Table. Maple Syrup Urine Disease Phenotypes). [3][13] There are 5 main clinical phenotypes of MSUD, based primarily on the residual BCKAD enzymatic activity that remains.[13]
Classic Maple Syrup Urine Disease
Classic MSUD is not only the most common subtype of the disorder but also the most severe and dangerous. Characteristic clinical features include:
- There is little to no BCKAD enzyme activity (0% to 2%).
- This type presents during the first 10 days of life.
- Maple syrup odor is first noticeable in the ear cerumen 12 hours after birth and in the urine by the end of the first week, which accelerates after protein ingestion.
- Without treatment, the course of classic MSUD is rapidly progressive and often fatal.
- Ketonuria is detectable 48 to 72 hours after birth, along with the first nonspecific clinical signs of lethargy, vomiting, irritability, and poor feeding.
- Encephalopathy develops 2 to 3 days later with worsening lethargy and intermittent apnea.
- Other symptoms include weight loss, seizures, and muscle tone variability. By 7 to 10 days after birth, coma and central respiratory failure may develop.
- If the MSUD remains untreated, death may follow in 2 months or less.
- Patients with the classic type of MSUD are particularly prone to exacerbations during periods of physical stress, such as with an infection, after surgery, or when fasting or with high protein consumption.
- Serum leucine titers are usually 2 to 4 times normal levels but can be much higher.
- Mutated genes comprise BCKHDA, BCKHDB, and DBT.
- Biochemical findings
- Elevated alloisoleucine
- Elevated plasma BCAA (ie, isoleucine, leucine, and valine)
- Elevated urine branched-chain ketoacids
- Ketonuria
- Positive urine dinitrophenylhydrazine test
Intermediate Maple Syrup Urine Disease
Intermediate MSUD is a much milder form, typically presenting between 5 months and 7 years of age. Characteristic clinical features include:
- These patients generally have some BCKAD activity remaining (3% to 30%).
- Symptoms include poor growth, developmental delays, poor intellectual progress, and decreased appetite with anorexia.
- The patient may develop encephalopathy and ketonuria when stressed by an infection, dehydration, high protein meals, or fasting.
- Maple syrup odor is noticeable in the ear cerumen and urine.
- Mutated genes comprise BCKHDA, BCKHDB, and DBT.
- Biochemical findings are the same as the classic subtype but are quantitatively less severe.
Intermittent Maple Syrup Urine Disease
Intermittent MSUD is a rare form of this uncommon disorder. While there is no restrictive age range for its initial presentation, it often appears within the first 1 to 2 years of life. Characteristic clinical features include:
- Residual BCKAD activity is 5% to 20% of normal.
- Regardless of when it first presents itself, it can progress rapidly.
- Initial symptoms may include seizures, encephalopathy, and ataxia.
- When physically stressed, the symptoms of affected patients can become much more severe.
- Mutated Genes: BCKHDA, BCKHDB, and DBT.
- Biochemical findings
- Normal branched-chain amino acid levels
- Similar profile to the classic subtype during disease exacerbation episodes
Thiamine-Responsive Maple Syrup Urine Disease
Thiamine-responsive MSUD is distinct from the other types of MSUD because patients with this variant respond well to large therapeutic doses of thiamine and are otherwise similar to the other forms of MSUD. Characteristic clinical features include:
- Residual BCKAD activity is 2% to 40% of normal.
- Mutated genes include DBT.
E3-Deficient Subtypes of Maple Syrup Urine Disease
E3-deficient subtypes of MSUD occur due to mutations in the gene encoding the E3 (ie, dihydrolipoamide dehydrogenase) subunit. Characteristic clinical features include:
- Patients usually have combined deficiencies of the E3 subunit and pyruvate/alpha-ketoglutarate dehydrogenase complexes.
- Clinically, patients present in the newborn period with lactic acidosis, encephalopathy, feeding difficulties, liver failure, and early death.
- This phenotype is associated with elevated lactate, alanine, and alpha-ketoglutarate levels.
Evaluation
Since MSUD is often rapidly progressive and can cause permanent harm within the first few weeks of life, it has become routine to perform diagnostic testing for MSUD on all newborns within 48 hours of birth. If this is missed, omitted, or unavailable in a timely fashion, the diagnosis of MSUD will depend on the prompt identification of the early clinical characteristics by a physician. In addition to the clinical features, biochemical analysis, and molecular testing play a significant role in disease confirmation and phenotype evaluation.
Initial blood screening for MSUD in neonates is routine and required in the United States and elsewhere; however, the screening may miss intermittent MSUD or genetic variants. Children and newborns should receive additional confirmatory testing for MSUD if any of the following are present, regardless of any initial negative neonatal screening tests:
- Sporadic encephalopathy and ketoacidosis
- Intermittent encephalopathy and ketoacidosis
- Encephalopathy and ketoacidosis during illness, trauma, or fasting
Prenatal Diagnostic Testing
Diagnosis requires mutational analysis to measure BCKAD enzyme activity in cultured chorion villus cells or amniocytes. BCAA acid concentrations can also be measured in amniotic fluid. Preimplantation diagnosis requires the identification of familial pathogenic variants. The preferred diagnostic method is molecular analysis.
Newborn Screening and Biochemical Evaluation
Since 1964, routine newborn screening has been performed for MSUD in the United States. The screening is best performed within 24 to 48 hours after birth using tandem mass spectrometry amino acid profiling. The process examines the fisher ratio and concentrations of leucine-isoleucine as a standard measure.[3] However, screening can give false-positive results in patients with isolated hydroxyprolinemia. Additional laboratory studies must be performed if elevated branched-chain amino acids are detected.[3]
Tandem mass spectrometry readily detects classic MSUD. However, it may not detect milder forms of MSUD due to normal leucine levels. Tandem mass spectrometry does not possess the capability to distinguish amino acids with the same mass (eg, hydroxyproline, leucine, isoleucine, and alloisoleucine).[3] Consequently, tandem mass spectrometry cannot differentiate hydroxyproline, leucine, and isoleucine. In such cases, plasma amino acid testing or a second-tier test, such as liquid chromatography, must be conducted to determine serum alloisoleucine levels. Such testing can reliably distinguish clinically significant MSUD from hydroxyprolinemia, which is harmless.
Plasma amino acid testing is the most critical and reliable diagnostic test for MSUD. This diagnostic modality is used to assess elevated levels of BCAAs and alloisoleucine. Elevated alloisoleucine levels >5 mmol/L confer high specificity and sensitivity for the diagnosis and are considered pathognomonic.[18] Even with very high serum leucine levels, plasma alloisoleucine is not detectable until the patient is 6 days of age.[18]
Additional laboratory studies include the dinitrophenylhydrazine (DNPH) test, gas chromatography, liquid chromatography, BCKAD enzyme activity, and molecular testing.[3] The urinary organic acids can be detected using gas chromatography-mass spectrometry, the dinitrophenylhydrazine (DNPH) test, and urine test strips. Gas chromatography-mass spectrometry detects branched-chain ketoacids, further supporting the diagnosis of MSUD. Branched-chain ketoacids usually follow after BCAA elevation and are detectable 48 to 72 hours after birth.[3] The DNPH test can also detect branched-chain ketoacids in the urine. The DNPH reagent and urine are mixed in equal volumes. The sample is then observed for 10 minutes for precipitation and color changes. Clear urine with no precipitate demonstrates a zero or negative score, while a yellow-white precipitate with opaque urine scores a 4 (ie, positive). The DNPH test's primary advantage stems from its ability to detect urinary branched-chain ketoacids quickly in an outpatient setting. Standard urine test strips enable the detection of ketonuria. This offers an alternative diagnostic methodology in settings with restricted access to more advanced laboratory testing. Ketonuria is considered a surrogate marker for possible MSUD.[13] Neonates suspected of MSUD should never be given a dietary "protein challenge," not only due to its potential danger but also because modern laboratory methods make it unnecessary. BCKAD enzyme activity can be measured in lymphoblasts, skin fibroblasts, and liver biopsy cells. In vivo measurements of BCKAD activity are not clinically useful because the activity levels do not correlate with leucine tolerance and oxidation.[13]
The molecular tests available to determine the 3 biallelic pathogenic gene variants include the BCKDHA gene which encodes the E1-alpha subunit of the BCKAD enzyme complex (MSUD Type 1A), BCKDHB gene which encodes the E1-beta subunit of the BCKAD enzyme complex (MSUD Type 1B), and the DBT gene which encodes the E2 subunit of the BCKAD enzyme complex (MSUD Type 2).[13] Genetic testing allows for a better understanding of the prognosis and genetic counseling of the family. Furthermore, molecular tests allow for an accurate assessment of the deficient BCKAD subunit. This helps determine the individualized therapies. There are over 190 pathogenic variations in BCKAD enzyme subunits. All detected variants are homozygous or compound heterozygous.
Diagnostic Approach
For certain patient presentations, varying diagnostic strategies are recommended.
Symptomatic adults
In symptomatic adults, diagnostic evaluation should include the following:
- DNPH testing can be used for the detection of alpha-ketoacids in the urine.[13]
- Branched-chain ketoacids and other organic acids can be detected using gas chromatography-mass spectrometry.
- The most definitive, informative, and reliable test is the identification of elevated serum alloisoleucine using plasma amino acid quantitative analysis.
Symptomatic newborns
In symptomatic newborns with a positive screening test or unexplained ketonuria, diagnostic evaluation should include the following:
- If the infants are older than 48 to 72 hours, screening tests such as DNPH and even common urine ketone test strips can be used.
- Plasma amino acid analysis to detect elevated BCAA and alloisoleucine.
- Gas chromatography-mass spectrometry is used to analyze the urine for ketoacids.
- Newborns and infants should not be challenged with higher than average protein intake.[13]
Newborns with an affected sibling
In newborns with a sibling already diagnosed with MSUD, diagnostic evaluation should include the following:
- If familial pathogenic variants are known, isolated blood from the umbilical cord can be used for variant detection by polymerase chain reaction (PCR), advanced sequencing, and melting analysis.
- If pathogenic variants are unknown, blood can be obtained from the umbilical cord to detect pathogenic variants.
- An amino acid profile indicative of MSUD should initiate dietary therapy. Molecular testing and urine organic acid analysis should follow.
- It is important to remember that DNPH can't be used as a screening test until 48 to 72 hours after birth.
Treatment / Management
Effective treatment of MSUD requires addressing the patient's nutritional needs and optimally managing acute metabolic decompensations.[19] A pediatric nutritionist and metabolic disease specialist should be involved in managing MSUD patients.
Medical Nutritional Therapy
Initiating nutritional therapy requires clinical confirmation or a positive newborn screening result. The mainstay of treatment remains the dietary restriction of branched-chain amino acids. These dietary modifications need to be maintained throughout life. Newborns who screen positive for MSUD should immediately be started on a special MSUD dietary formula with no BCAA without waiting for confirmatory testing. There is evidence that a high rate glucose infusion at 11 mg/kg and 20% intravenous lipids at a rate of 2 g/kg daily can be helpful in addition to the dietary therapy in limiting brain damage, even in newborn MSUD patients who are initially asymptomatic.[20] An insulin infusion may be helpful in some cases to promote anabolism. The goals of nutritional therapy are as follows [21]:
- Promote anabolism
- Prevent catabolism
- Promote average growth and weight gain
- Preserve intellectual function
- Restriction of branched-chain amino acids, especially leucine, while supplementing valine and isoleucine
- Maintain plasma BCAA levels within the required treatment ranges
- Evaluate thiamine responsiveness
The allowed amounts of dietary BCAA are titrated into the diet using biochemical lab values and growth measurements during respective life periods. Long-term treatment warrants accurate assessment of caloric needs and BCAA restriction with valine and isoleucine supplementation.[3] BCAA-free medical foods can provide up to 80% to 90% of protein needs. Valine and isoleucine help promote anabolism. Leucine supplementation is usually unnecessary because it is found in ample amounts in breast milk or formula. Favorable intellectual outcomes can be achieved if leucine concentrations are maintained between 75 and 300 mmol/L.[22] Plasma valine and isoleucine levels should also be maintained between 200 to 400 mmol/L.[21] Thiamine should be supplemented at a dose of 50 to 200 mg/day in all patients with MSUD for 4 weeks. In thiamine-responsive patients, this thiamine supplementation should be continued. Thiamine should not be supplemented in patients diagnosed with the homozygous 1312T>A mutation, common in Mennonites, or with mutations leading to <3% of BCKAD activity. Administration of sodium chloride may help maintain serum sodium concentrations and plasma osmolarity and can help prevent the development of cerebral edema or even a potentially fatal brain herniation. Carnitine supplementation may combat some oxidative stress caused by elevated BCAA and improve neurocognitive outcomes.[1]
Treatment of Acute Metabolic Decompensation
Metabolic decompensations, typically at plasma leucine levels >380 mmol/L, usually occur due to dietary noncompliance or infections. Dietary noncompliance raises the BCAA levels but rarely progresses to decompensation and encephalopathy without additional physical or metabolic stress. However, trauma and infections can trigger protein catabolism, leading to a metabolic crisis. Decompensation arises more commonly in the first year of life and after age 15.[23] Strauss et al. indicated that vomiting and viral gastroenteritis are the most common cause of hospitalization that triggers an acute decompensation episode in patients with MSUD.[11] Other common causes of hospitalization include viral bronchiolitis, sinusitis, neonatal encephalopathy, and urinary tract infections.[11]
Residual BCKAD activity and liberation of leucine from catabolism determine the risk of metabolic crisis.[13] Patients with a higher residual BCKAD activity have a better tolerance for leucine. Furthermore, during an illness, these patients face less severe elevations in leucine. The main aim of therapy is to suppress protein catabolism and promote protein synthesis.[3] Management strategies in more severe cases include:
- Treating the underlying stressor causing the metabolic crisis
- Restricting protein intake for 24 to 72 hours
- Providing ample caloric support
- Providing adequate hydration to maintain metabolic homeostasis
- Providing supplementation with cofactors
- Eliminating toxic metabolites
- Treating associated clinical sequelae
- Correcting metabolic abnormalities
- Using dialysis (peritoneal and hemodialysis) in the most severe or acute cases refractory to other treatment
Home Therapy
Dried blood spot evaluations of amino acid concentrations can be done routinely for home monitoring of less severe cases. Once or twice a week is suggested for infants. Older children and adults should be checked every 1 to 2 weeks.[13] Home healthcare personnel and family members can be instructed to use a dinitrophenylhydrazine reagent regularly to detect high urine branched-chain ketoacids or when an exacerbation is suspected. This allows for the timely detection and home management of mild to moderate cases of acute metabolic decompensation. Experienced clinicians can help manage dietary leucine restrictions, sick day formulas, and close outpatient monitoring in such cases.[13]
Specifically, sick day protocols require a 120% increase in BCAA-free amino acid formula intake, fluid administration of 150 mL/kg body weight, a 50% to 100% decrease in leucine intake, and frequent small feedings.[3] Amino acid transport into the brain will remain permanently impaired, and there will be a chronic deficiency of cerebral neurotransmitters (eg, dopamine, glutamate, tryptophan, serotonin, and γ-aminobutyric acid). Ensuring that nutritional treatment is aggressive and provides sufficient energy is vitally important. Dietary therapy for MSUD is lifelong.
Inpatient Therapy [13]
Inpatient management for MSUD is more intensive compared to outpatient management. In addition to dietary strategies, clinicians should effectively treat the underlying stressor (eg, fever, dehydration, infection, surgery, and inflammation). Nausea and vomiting should be controlled with antiemetics. Initially, leucine concentration should be reduced by 750 mmol/L or more every 24 hours, which can be achieved via insulin and glucose infusions. Ideally, leucine levels should be maintained from 200 to 300 mmol/L.
Upon clinical improvement, total parenteral nutrition can reintroduce protein into the diet at 25% to 50% of normal intake. Depending on the clinical situation, this intake can be increased over the next few days. The estimated energy requirement (EER) must be provided at least 1.25 times the neonates' weight or surface area. The EER is the average dietary energy intake estimated to maintain a healthy energy and nutritional balance in an individual with good health based on age, gender, weight, height, and physical activity level. In adults and older children, increasing caloric intake to 3 times the EER, typically around 6,000 calories per day, may be necessary to prevent catabolism.[24] This may require central venous access. Lipids should constitute 40% to 50% of the total calorie intake. Dextrose 25% IV solutions should be used to maximize caloric needs while minimizing possible hypervolemia. A continuous insulin infusion of 0.02 to 0.15 units/kg/hour can optimize blood glucose levels at between 100 and 160 mg/dL and promote anabolism. The total protein equivalent intake as BCAA-free amino acids should be 2.0 to 3.5 g/kg body weight daily, including both oral and parenteral intake.
Total isoleucine and valine supplements should be 20 mg to 120 mg/kg daily. The plasma concentration goal is between 400 and 800 mmol/L. The use of BCAA-free intravenous solutions in emergency settings or early MSUD decompensation appears to be safe and effective, particularly for pediatric patients and others where oral or enteric therapy is unavailable.[25] As parenteral BCAA-free amino acid solutions may be challenging to obtain, the use of continuous nasogastric delivery of a suitable MSUD formula of 0.7 to 1.2 Kcal/mL at 30 to 60 mL/H is suggested as an alternative dietary calorie source regardless of patient age.[24] This enteral feeding should be supplemented with 1% isoleucine and valine solutions to meet optimal nutritional protein goals.[24] Isoleucine and valine should be supplemented at 20 to 120 mg/kg daily. The intake of these supplements should be adjusted to maintain a steady plasma concentration of 400 to 600 mmol/L for each. Enteral tyrosine supplementation should be administered at 100 to 400 mg/kg daily to treat focal or generalized dystonia. Glutamine and alanine supplementation should be given at 150 to 400 mg/kg each daily.
Nutritional goals can typically be achieved by combined parenteral and enteral feeding. Sodium levels should be maintained within the physiological range. Underlying acid-base disturbances should also be corrected. Osmolarity fluctuations of >5 mOsm/L daily should be avoided while maintaining urine output. Experts also recommend preventing and treating hypokalemia and hypophosphatemia associated with intravenous glucose and insulin therapy. The use of glucocorticoids and vasoactive catecholaminergic agents should be limited. Hemodialysis and peritoneal dialysis can also be used to rapidly correct BCAA and excess ketoacids during an episode of acute decompensation if needed.[13] Sodium phenylbutyrate is a nitrogen scavenger used primarily to treat urea cycle disorders; it can also reduce branched-chain amino acid levels, making it useful in patients with intermediate MSUD.[26]
Suggested Intensive Care Unit Monitoring
For patients being cared for in the intensive care unit with an acute exacerbation, laboratory monitoring should include:
- Serum glucose every 4 to 6 hours
- Serum osmolality and electrolytes every 6 to 12 hours
- Plasma amino acids, serum phosphorus, and magnesium every 12 to 24 hours
- Serum lipase, amylase, and transaminases every 24 to 48 hours [24]
Orthotopic Liver Transplantation
The liver is responsible for expressing 10% of BCKAD activity. Restoring 9% to 13% of the normal BCKDH enzyme activity will effectively control branched-chain amino acid metabolism.[7] Therefore, liver transplantation is recommended for classic (severe) MSUD patients who cannot be managed through diet. Most commonly, the liver from an undeceased and unrelated individual is used.[3] Posttransplantation, the residual activity of BCKAD can rise to mild MSUD. Transplantation essentially eliminates the need for severe dietary restrictions and helps avoid episodes of metabolic decompensation, although it does not reverse previous brain damage, cognitive dysfunction, or psychiatric illnesses.[27] However, transplantation does prevent the progression of further symptoms.[28][29] If performed promptly after initial diagnosis, reported liver transplantation outcomes suggest minimal, if any, long-term neurological impairment in these patients.[29] Liver transplant indications include:
- Psychomotor disabilities
- Poor metabolic control
- Frequent metabolic decompensations
- Poor quality of life
- Classic (severe) MSUD
Management in Pregnancy
It is indeed possible for women with MSUD to deliver a healthy child. The mother must be educated about the potential teratogenic risks of elevated maternal leucine concentration. In such patients, tight metabolic control before and throughout gestation is critical. As the placenta and fetus develop, the maternal need for protein and BCAA exponentially increases. Therefore, periodic plasma amino acid concentrations and fetal growth measurements are imperative to avoid possible nutritional deficiencies. Maintaining BCAA levels between 100 and 300 mmol/L is compatible with normal infant delivery. At delivery, a referral to an experienced metabolic center should be considered. This is because the postpartum period is deemed particularly dangerous for the mother. Events such as labor stress, internal blood sequestration, and uterine involution can act as a source of metabolic decompensation. Therefore, extra measures must be taken to counteract catabolism during the immediate postpartum period.
Management of Other Maple Syrup Urine Disease Complications
In addition to advising patients never to exceed their daily allowed dietary branched-chain amino acids and follow their recommended monitoring guidelines as a preventative intervention, clinicians should manage various complications that may develop with MSUD.
Cerebral edema
Significant cerebral edema from MSUD can begin during the first week of life.[30] Initial signs are typical for MSUD and include poor feeding, vomiting, irritability, lethargy, and periods of apnea, which worsen as the encephalopathy progresses. Left untreated, this can rapidly progress, leading to seizures, coma, respiratory failure, and death.[20] Cerebral edema in MSUD is due to increased branched-chain amino acids in the brain and deficient levels of other essential amino acids and neurotransmitters (glutamate and tryptophan).[15][30] The 25% IV dextrose, used to maintain caloric needs in MSUD patients, increases renal leucine excretion but also causes sodium wasting by the kidney. This urinary sodium loss can worsen cerebral edema.[31]
Ultrasound cranial imaging can be used in neonates to help detect cerebral abnormalities but is very operator-dependent. MRI is the imaging procedure of choice for detecting cerebral edema or brain herniations.[32] An MRI of the brain can show signs of edema and atypical signal density characteristic of MSUD.[33] This can progress, becoming more severe and localized. The sites most affected include the dorsal limb of the internal capsule, the cerebral peduncles, the dorsal brainstem, and the deep cerebellar white matter.[20] Acute suppression ratio in quantitative electroencephalography can be an early indicator of increased intracranial pressure, making it potentially a very useful predictor of cerebral edema in acute MSUD decompensations.[34]
Due to the potentially rapid progression of significant cerebral edema and encephalopathy, treatment in neonates should begin immediately after a positive MSUD screening without waiting for confirmatory testing. Initial treatment in newborns involves dietary therapy (a special formula that contains no branched-chain amino acids) along with IV administration of glucose and intravenous lipids, even in those babies without symptoms.[20] The head of the bed should be elevated, and a PICC line should be inserted to allow the delivery of nutrition without the need for excess fluid volume.[13] The administration of furosemide and mannitol, followed by 3% hypertonic saline, as needed, should be considered in patients with MSUD demonstrating evidence or progression of cerebral edema.[24] The following dosages of each agent are typically used:
- Furosemide 0.5 to 1.0 mg/kg body weight per dose
- Mannitol 0.5 to 1.0 g/kg body weight per dose
- Titrate 3% hypertonic saline at a rate of 2 to 3 mEq/kg per dose OR a saline drip at 2 to 10 mEq/kg per day based on the following levels:
- Serum osmolality 285 to 300 mOsm/kg of water
- Serum sodium 138 to 145 mEq/L
- Serum osmolality change <0.2 mOsm/kg of water/hour (≤5 mOsm/kg of water/day)
Malignant cerebral edema is the most common immediate cause of death from MSUD, which can be as high as 25%.[24] When treated promptly and correctly, the cerebral edema slowly resolves over the next several months, but often not without some loss of brain substance or periventricular white matter disease.[20] See the companion StatPearls article "Cerebral Edema" for information on managing cerebral edema.[35]
Cerebral herniation
Initial signs of impending cerebral herniation from increasing cerebral edema include vomiting, dilated pupils, worsening lethargy, hypotension or hypertension, seizures, breathing irregularities, abnormal body posturing or muscular activity, altered mental status, abnormal reflexes, unconsciousness, coma and death from respiratory and cardiac arrest. In adults, patients may have an altered mental status and complain of nausea or headache. The diagnosis is confirmed by imaging.[36] See the companion StatPearls article "Brain Herniation" for additional information.[37] Treatment is based on medical management to reduce cerebral edema and intracranial pressure, including:
- Elevate the head.
- Induce hyperventilation with the help of a face mask or endotracheal tube.
- Restrict fluid intake but avoid dehydration.
- Infuse mannitol, furosemide, and 3% hypertonic saline to reduce cerebral edema.[24]
- Transfer the patient immediately to a pediatric/neonatal intensive care unit.
- Consider hypothermia, which slows down brain metabolism but increases the risk of cardiac arrhythmias and infections.
- Consider induced pentobarbital coma, which can reduce cerebral blood flow but may cause hypotension.
- Avoid decompressive hemicraniectomy; though tempting, when used in traumatic cases of cerebral herniation, a recent study indicated it offered only a minimal survival advantage but resulted in more patients left in a persistent vegetative state and more significant disabilities than medical therapy alone.[38]
Infection
Patients with MSUD are predisposed to candida infections and catheter-based bacterial or fungal infections. Monitoring patients for hospital-acquired infections continuously is vital. Failure to treat infections aggressively can result in acute metabolic decompensation.
Acute pancreatitis
During an episode of acute metabolic decompensation, the patient may develop symptoms of epigastric or mid-back pain, anorexia, or vomiting. This should raise suspicion of acute pancreatitis and immediately indicate the need for serum lipase and amylase levels. At that point, enteral feeding should be suspended, and the patient should be kept NPO. Treatment is usually supportive, and the patient's nutritional needs can be managed using special parenteral solutions.
Neuropsychiatric illnesses
Adult and adolescent patients are at a significantly increased risk of developing anxiety, depression, panic disorder, and ADHD. Standard antidepressants and psychostimulant drugs are typically effective in these patients.
Secondary complications
Elective surgical procedures should be planned in coordination with a metabolic specialist.[13] Emergency procedures and trauma care are likely to provoke an exacerbation or decompensation of MSUD, and a metabolic specialist in the disorder should be involved early in the care of these patients.[13]
Differential Diagnosis
Clinicians should exclude other clinically distinct entities that can also manifest as neonatal encephalopathy. These include β-ketothiolase deficiency, birth asphyxia, encephalitis, β-hydroxy β-methylglutaryl-CoA lyase deficiency, hypoglycemia, kernicterus, meningitis, nonketotic hyperglycinemia, organic acidopathies such as propionic acidemia and methylmalonic acidemia, status epilepticus, urea cycle defects.[13] Sotolone, an isoleucine metabolite that is also found in fenugreek, some aged rum, molasses, candy cap mushrooms, caramel, sherry, and lovage, is responsible for the characteristic maple syrup odor in cerumen and bodily secretions.[9] Excess ingestion of these items during pregnancy can result in a false diagnosis of MSUD. In NICUs, topical benzoin can also give off a similar sweet odor.[13]
Prognosis
A good prognosis can be expected in patients who begin therapy before or immediately after developing symptoms. Plasma leucine concentrations and the severity and duration of cerebral edema and encephalopathy affect neurocognitive outcomes. Patients with classic MSUD can show higher school performance than neurocognitive testing would predict.[39] 61% of adult patients with MSUD live independently and integrate well into society.[23] However, 56% of patients still require psychological and psychiatric care.[23] Patients with late-onset MSUD can suffer from slight developmental delays depending on the residual BCKAD activity.[13] A delayed diagnosis of more than 7 to 14 days in classic MSUD can result in an irreversible learning disability, coma, respiratory failure, brain damage, seizures, and cerebral palsy.[13] The development of metabolic acidosis during an acute episode appears to be associated with a poorer outcome.[40] The mortality from MSUD during an acute episode has been estimated as high as 25%. The most common cause of death is malignant cerebral edema.[24]
Complications
Failure to diagnose and treat MSUD in a time-sensitive manner can result in serious consequences. Patients undergoing a treatment plan can develop an acute illness resulting in a sudden increase in levels of branched-chain amino acids. This metabolic crisis is usually indicated by the development of clinical symptoms such as extreme fatigue, irritability, vomiting, and loss of alertness. If the patient remains undiagnosed or untreated, the following complications can arise:
- Acute pancreatitis
- Blindness
- Cerebral edema
- Cerebrovascular ischemia
- Essential amino acid deficiency presenting as anemia, hair loss, growth failure, and acrodermatitis
- Intellectual disabilities
- Irreversible neurological damage
- Metabolic acidosis
- Muscle spasticity
- Osteoporosis
- Recurrent esophageal candidiasis due to T-cell suppression from elevated plasma leucine.
- Seizures
Deterrence and Patient Education
A thorough understanding of the disease will ensure adherence to optimal patient care strategies. Furthermore, it allows physicians and other healthcare team professionals to provide the best patient outcomes based on evidence-based medicine. Educational resources include:
- Educational handouts obtained online from the New England Consortium of Metabolic Programs
- The American College of Pediatrics website
- MSUD family support group
- National Library of Medicine Genetics Home Reference
- NCBI genes and diseases
- Metabolic Support UK
- European Registry and Network for Intoxication Type Metabolic Diseases
- Organic Acidemia Association
Pearls and Other Issues
Key facts that clinicians should bear in mind include:
- MSUD is genetically transmitted in an autosomal recessive manner. Each sibling of an affected patient has a 25% chance of having the disease, a 50% chance of being an unaffected carrier, and only a 25% chance of being both unaffected and not a carrier.
- Carrier testing in blood relatives is recommended if a family member has been diagnosed. Prenatal diagnosis for pregnant blood relatives is suggested.
- In suspected cases of MSUD, testing serum acylcarnitines and lactate concentration is recommended to eliminate several similar conditions, including dihydrolipoamide dehydrogenase and ketothiolase deficiency.[13]
- The most definitive laboratory diagnostic test for MSUD is a plasma amino acid quantitative analysis finding an alloisoleucine level of more than 5 mmol/L.[18] Elevated serum BCAA will be detectable in the plasma within the first 24 hours, but alloisoleucine will not be detectable before 6 days of life.
- MSUD is not associated with either hypoglycemia or hyperammonemia.[13]
- Serum leucine levels do not always correlate with brain levels.
- Dietary treatment of MSUD is a lifelong therapy.
- The individual states in the US determine required newborn testing and may comprise 50 or more rare congenital disorders, including:
- Congenital adrenal hyperplasia
- Cystic fibrosis
- Galactosemia
- Homocystinuria
- HIV
- Maple syrup urine disease
- Phenylketonuria
- Primary congenital hypothyroidism
- Sickle Cell
- Thalassemia
- Gene therapy offers much promise in correcting the defect that causes MSUD, but the extreme rarity of the disorder makes it unlikely any such treatment will be available in the foreseeable future.
Management Summary
Critical aspects of MSUD management include:
- Neonates who have an initial MSUD screening test that is positive should be treated immediately and aggressively without waiting for confirmatory tests or symptom development.
- Immediate treatment includes MSUD dietary formula, IV glucose, parenteral lipids, and possible insulin infusions.
- Renal replacement therapy, including hemodialysis, peritoneal dialysis, and continuous kidney replacement therapies, have been used in acute decompensations, and short-term safety has been demonstrated even in infants.[41]
- Imaging (CT or MRI) is necessary to identify potentially life-threatening cerebral edema and brain herniation.
- Treatment of cerebral edema and brain herniation is primarily medical, not surgical, with furosemide, mannitol, and 3% hypertonic saline.
- Confirmatory testing for MSUD is most reliably done by plasma amino assay analysis, which cannot be performed before 6 days after birth.
- Any neonatal or pediatric patient with unexplained encephalopathy, together with ketoacidosis, should be tested for MSUD even if the original neonatal screening was negative.
Enhancing Healthcare Team Outcomes
Optimal management of maple syrup urine disease requires an interprofessional team consisting of an internist, pediatrician, geneticist, metabolic specialist, pediatric nutritionist, social worker, and pediatric nurse. The mainstay of therapy is medical and nutritional therapy. The healthcare team must collaborate to assess caloric needs and execute dietary treatment strategies such as BCAA restriction, BCAA-free amino acid ingestion, and supplementation of valine and isoleucine.
The pediatric nutritionist must educate the patient never to exceed the daily intake of amino acids. They should also advise the patient to adhere to a lifelong restricted diet. The geneticist must counsel the patient or their parents about the implications of their inherited disorder to help them make informed medical and personal decisions. Given the high incidence of neurological and psychological symptoms, both neurologists and psychiatrists may need to be heavily involved. Treatment is ongoing and lifelong, and new developments are possible. Therefore, a patient should be closely followed throughout childhood into adulthood with appropriate care transitioning. Early diagnosis, aggressive treatment, and controlling plasma leucine levels help improve neurological outcomes.