Continuing Education Activity
Molecular diagnostics encompasses the analysis of human, viral, and microbial genomes and the products they encode. Molecular genetics utilizes molecular biology's laboratory tools to relate genetic structure to protein function and, ultimately, health and disease. Variants identified during genetic testing are classified based on diverse evidence types, as the American College of Medical Genetics and Genomics recommends, emphasizing the need for board-certified geneticists to interpret the results. Integrating genetic testing methodologies with clinical expertise is crucial in translating molecular genetics advancements to better patient care.
The field of molecular genetic and genomic testing is undergoing rapid change due to improvements in our understanding of the molecular causes of uncommon and common illnesses and DNA analysis technologies. The advent of molecular genetics has revolutionized healthcare by offering unprecedented insights into the genetic basis of diseases, enabling personalized diagnostics, treatment strategies, and risk assessments. However, this progress brings with it the responsibility for healthcare providers to stay updated with the latest advancements and best practices in genetic testing.
This activity for healthcare professionals is designed to enhance learners' proficiency in identifying patients with indications for molecular genetics testing and interpreting genetic test results. Participants acquire a broader grasp of specimen collection, procedures, indications, potential diagnosis, normal and critical findings, interfering factors, and complications. Learners gain insights into the complexities of molecular genetics, preparing them to collaborate with an interprofessional team that aims to improve outcomes for patients who need molecular genetics testing.
Objectives:
Identify clinical encounters appropriate for genetic molecular testing, distinguishing cases where such testing can contribute to diagnosis, prognosis, or treatment decisions.
Evaluate genetic test results accurately, discerning their clinical significance and relevance to patient management.
Differentiate between genetic testing methodologies, understanding their strengths, limitations, and optimal applications to diagnose patients.
Implement best interprofessional collaboration and communication practices to ensure that patients who need molecular genetics testing receive comprehensive care that considers their medical, psychological, and social needs, thus improving outcomes.
Introduction
Molecular genetics testing is fundamental in evaluating inherited disorders, somatic or acquired diseases with genetic associations, and pharmacogenetic responses. Genotyping can provide valuable disease diagnosis, prognosis, and progression indicators, guide treatment selection and response, and identify gene-specific therapeutic targets.[1] Human genetic material primarily consists of double-stranded, helical DNA. This molecule has a backbone composed of alternating sugar (deoxyribose) and phosphate groups, with hydrogen bonds linking nitrogenous base pairs. Specifically, adenine (purine) pairs with thymine (pyrimidine), while guanine (purine) pairs with cytosine (pyrimidine), forming the complementary base pairs within the DNA double helix.[2][3]
DNA in human cells is wrapped around histone proteins and packaged into nucleosome units, compacted further to form chromosomes.[4] Somatic cells normally have 23 chromosome pairs, with 1 pair comprised of the sex chromosomes X and Y. Each chromosome has DNA with a terminal stretch of short repeats called “telomeres” and additional repeats in the centromere region.[5]
Humans have 2 sets of 23 chromosomes, one derived from the mother’s egg and the other from the father's sperm. Therefore, each egg and sperm is a single or haploid set of 23 chromosomes. Combining the 2 creates a diploid set of human DNA, allowing each individual to possess 2 different sequences, genes, and alleles on each chromosome.[6] Homologous recombination during meiosis generates unique allele combinations in gametes, leading to genetic diversity among offspring in the human population.[7]
The complete decoding of the human genome sequence and the development of powerful identification and cloning methods for genes linked to inherited diseases have transformed the practice of molecular genetics and molecular pathology. Advanced molecular analysis methods can now determine presymptomatic individuals' illness risk, detect asymptomatic recessive trait carriers, and prenatally diagnose conditions not yet evident in pregnancy.[8] Molecular genetics techniques are often the only approaches to these puzzles. Thus, genetic tests are powerful tools for diagnosis, genetic consultation, and prevention of heritable diseases.[9]
Many genetic tests can analyze gene, chromosome, and protein alterations. A clinician often considers several factors when selecting the appropriate test, including suspected conditions and their possible genetic variations. A broad genetic test is employed when a diagnosis is uncertain, while a targeted test is preferred for suspected specific conditions.[10] Molecular tests look for changes in 1 or more genes. These tests analyze the sequence of DNA building blocks (nucleotides) in an individual's genetic code, a process known as DNA sequencing, which can vary in scope.[11]
The targeted single variant test identifies a specific variant in a single gene known to cause a disorder, eg, the HBB gene variant causing β-globin abnormalities that give rise to sickle cell disease. This test assesses the family members of an individual with the known variant to ascertain if they have the familial condition.[12] Single-gene tests examine genetic alterations in 1 gene to confirm or rule out a specific diagnosis, notably when many variants in the gene can cause the suspected condition. Gene panel tests look for variants in multiple genes to pinpoint a diagnosis when a person has symptoms that may fit various conditions or when many gene variants can cause the suspected condition.[13][14]
Whole-exome sequencing or whole-genome sequencing tests analyze the bulk of an individual's DNA to find genetic variations. This approach is useful when a single-gene or panel testing has not provided a diagnosis or when the suspected condition or genetic cause is unclear.[15] This sequencing method is often more cost- and time-effective than performing multiple single gene or panel tests.[16]
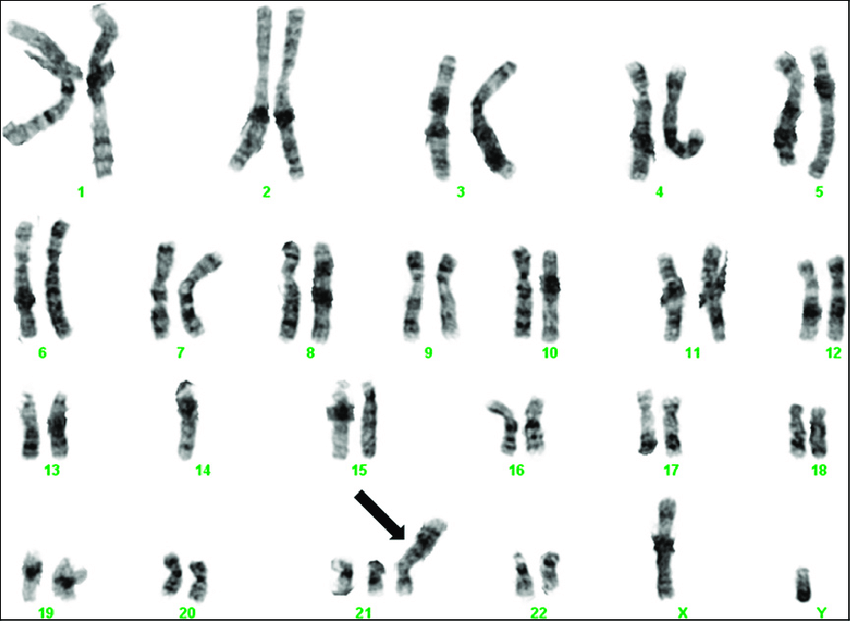
Chromosomal tests analyze whole chromosomes or long DNA lengths to identify significant alterations, including extra or missing chromosome copies (trisomy or monosomy), large chromosomal segment duplications or deletions, and segment rearrangements (translocations) (see Image. Trisomy 21 on G-Banded Chromosomal Studies).[17] Chromosomal tests are employed when specific genetic conditions linked to chromosomal changes are suspected. For instance, Williams syndrome results from deleting a chromosome 7 segment.
Gene expression tests assess gene activation status in cells, indicating whether genes are active or inactive, with activated genes producing mRNA molecules that serve as templates for protein synthesis.[18] The mRNA produced helps determine which genes are highly active. Too much activity (overexpression) or too little activity (underexpression) of specific genes may suggest particular genetic disorders, including various cancer types.[19] Biochemical tests assess protein or enzyme levels and activity rather than directly analyzing DNA.[20] Abnormalities in these substances may indicate DNA changes underlying a genetic disorder.
Heritable mutations are detectable in all nucleated cells and are thus considered germline or constitutional genetic changes. Somatic genetic changes are characteristic of acquired or sporadic diseases like cancer.[21] Both scenarios are investigated using similar molecular biology methods to detect DNA and RNA variations, although the interpretation and utility of the laboratory results often differ significantly.[22]
Fluorescent in situ hybridization (FISH), chromosomal microarray analysis (CMA), and cytogenetic analysis (karyotyping) can be used to detect gross mutations like whole- and large-scale gene deletions, duplications, or rearrangements. Conventional karyotyping identifies rearrangements over 5 DNA megabases.[23] FISH has a resolution of 100 kilobases to 1 megabase. Minor alterations, such as single-base substitutions, insertions, and deletions, are detectable with single-strand conformation polymorphism (SSCP) and sequence analysis through next-generation sequencing (NGS). NGS uses genomic DNA (gDNA) or complementary DNA (cDNA) and has 3 modalities: whole genomic DNA, targeted, and exome sequencing.[24]
Denaturing high-performance liquid chromatography (DHPLC) can detect small deletions and duplications. Multiplex ligation-dependent probe amplification (MLPA) extends the range of deletions and duplications detected, bridging the gap between FISH or cytogenetic analysis and HPLC. MLPA is particularly useful in identifying complete or single and multiexon deletions or duplications.[25][26]
Specimen Collection
Peripheral blood is the specimen required for FISH, MLPA, DHPLC, and sequencing. Amniotic fluid cells and, more recently, cell-free fetal DNA may be used for noninvasive prenatal testing.[27] Ethylenediaminetetraacetic acid is the most commonly used anticoagulant for molecular-based testing. However, acid citrate dextrose (ACD) is an acceptable alternative in cases where cellular form and function must be preserved.
ACD A and ACD B are the only ACD tube designations recognized, differing only by their additive concentrations.[28] Both enhance white blood cell viability and recovery for several days after specimen collection, making them suitable for molecular diagnostic and cytogenetic testing.
Procedures
FISH utilizes fluorescent DNA probes to target specific gene sequences in interphase or metaphase cells, enabling their visualization and detection. Housekeeping gene probes always serve as positive internal controls. The probe must be large enough to hybridize specifically with the target without impeding the hybridization process. Conventional FISH involves pipetting the hybridization mix onto the cytological sample and incubating them together.
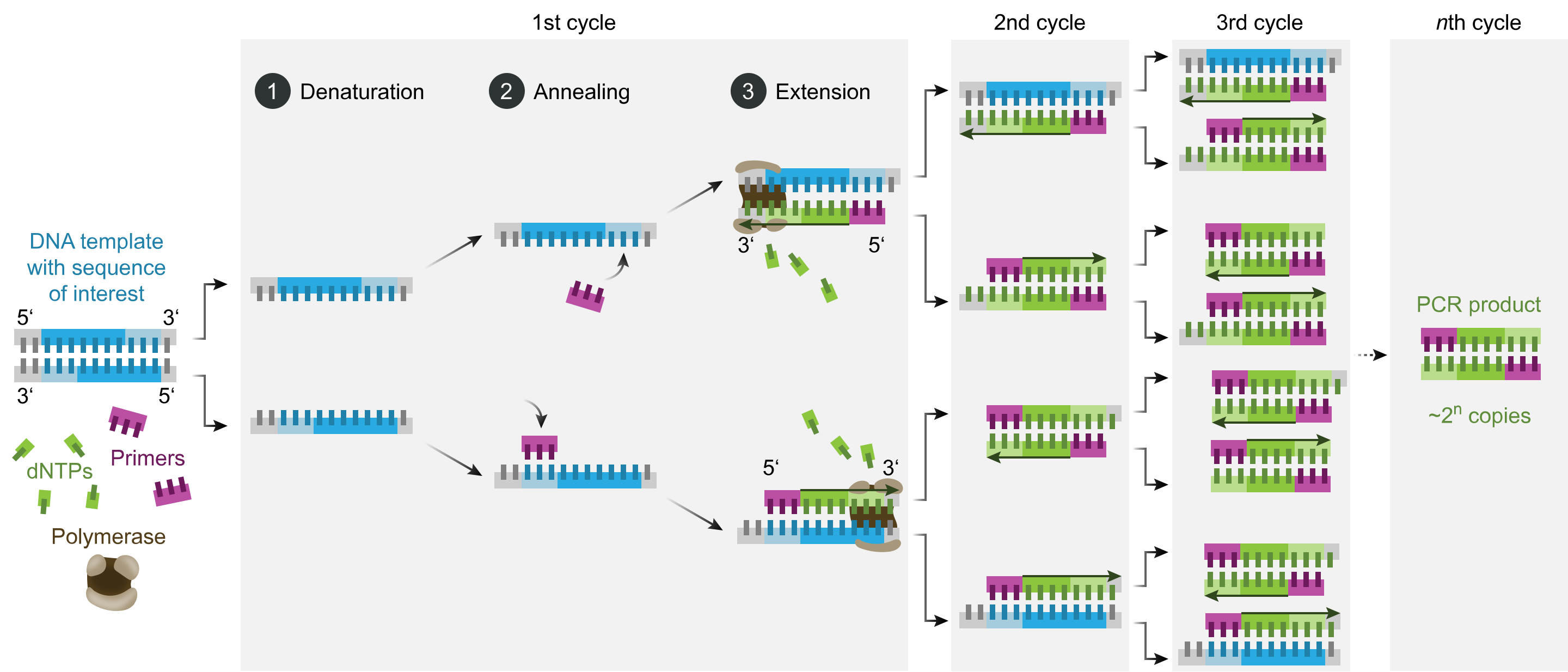
The technique can be applied to suspended cells, cultured cells, and frozen or formalin-fixed paraffin-embedded tissue sections, with subsequent cell sorting for fluorescence signal separation.[29] Preserving nucleic acid integrity and cell morphology is necessary during sample fixation. The experimental FISH procedure includes several preparatory steps, the hybridization reaction itself, and the removal of unbound probes.[30] The probe may be directly labeled with fluorophores or targeted for fluorescent detection using labeled antibodies or similar substrates. Different tags may be used, and different targets may be detected in the same sample simultaneously (multi-color FISH). Tagging is performed in various ways, including nick translation or polymerase chain reaction (PCR) using tagged nucleotides (see Image. Polymerase Chain Reaction). Probes can vary from 20 to 30 nucleotides to much longer sequences.
Locus-specific probes provide insight into gene amplification, deletion, or normal copy number status. Dual-fusion probes are adept at identifying frequently translocated gene regions associated with cancer development. These probes target regions spanning the breakpoints of translocation partners. Intact green and red signals are determined when they are closer than one signal's width. Conversely, a break in the gene sequence results in separate green and red signals.[31]
Break-apart probes target 2 areas of a specific gene sequence, using a green fluorescent label on one end and a red fluorescent label on the other. Intact gene sequences typically produce a yellow signal, known as a fusion signal. Whole-chromosome probes consist of smaller probes, each binding to different sequences along a chromosome. [32] Multiple probes, labeled with fluorescent dyes, enable unique color labeling of each chromosome, creating a spectral karyotype—a full-color chromosome map identifying all chromosome pairs.[33] Whole-chromosome probes are useful for examining chromosomal abnormalities, such as translocations.
Chromosomal microarray (CMA) consists of thousands of tiny probes, each representing small DNA fragments from known locations on the 46 chromosomes. CMA detects imbalances in chromosomal material between patient and control DNA samples, identifying copy number differences—whether gains (duplications) or losses (deletions)—in specific DNA segments.[34] These differences pinpoint the cause of the patient's health condition based on the location and type of change detected.[35]
Denaturing high-performance liquid chromatography (DHPLC) relies on differential chromatography retention of DNA heteroduplexes post-denaturation and renaturation. DNA heteroduplex migration is influenced by both molecule length and melting temperature, which is crucial for test sensitivity. DHPLC typically compares 2 PCR products amplified from 2 genes: 1 wild type and 1 mutated. These PCR products can originate from either RNA (cDNA) or genomic DNA. The PCR products are denatured at 95 °C and gradually reannealed by cooling from 95 °C to 65 °C before chromatography. A major advantage of this technology is that multiple samples can be pooled together for variant detection.[36] Sequencing detects single-base substitutions and small deletions and insertions in DNA fragments ranging from 80 to 1500 base pairs, with close to 100% accuracy within minutes.
When a mismatch is present, both the original homoduplexes and 2 heteroduplexes are simultaneously produced. The original homoduplexes form from the reannealing of perfectly matching sense and antisense strands (25% each). The heteroduplexes form from the reannealing of the sense strand of one homoduplex with the antisense strand of the other (also 25% each). Heteroduplexes denature more extensively than homoduplexes, resulting in earlier elution from the chromatography column. The separation of all 4 species is based on their differences in stacking interactions with the chromatography column (solid phase). More detailed theoretical explanations of DHPLC are available in the literature.[37]
MLPA utilizes genomic DNA samples, with specific MLPA probes hybridizing with denatured genomic DNA. These probes are uniquely designed to hybridize adjacent to each other on the target DNA region and confer a distinct length to each amplified MLPA probe pair. Detection and quantification occur via capillary electrophoresis.[38] All MLPA probes are amplified using the same primer pair, with the abundance of each fragment proportional to its target's copy number in the sample.
NGS amplifies DNA with random priming, providing a genome-wide view of the patient's genetic background through millions of reads. Library generation begins with nucleic acid fragmentation, representing the individual's entire genome or transcriptome. Whole-exome sequencing uses cDNA fragments, whereas the whole-genome modality includes complete genomic DNA. Fragments join using enriched sequence adaptors. Only some genes (gene panel) are analyzed in targeted libraries. Fragments hybridize with cDNA fragments for the region or genes of interest and are specifically enriched.[39] During sequencing, nucleotide addition is detected by fluorescent dyes or pH changes from hydrogen ion release during DNA polymerization.[40]
Sanger sequencing begins with PCR-based target DNA amplification, followed by removing excess deoxynucleotide triphosphates (dNTPs) and PCR primers. The Sanger method has 99.99% base accuracy and is thus the "gold standard" for validating DNA sequences, including those from NGS. The test's steps include denaturing the double-stranded DNA (dsDNA) into 2 single-stranded DNA (ssDNA), attaching a primer corresponding to one end of the sequence, and sequencing 4 polymerase solutions with 4 dNTPs. Only one type of ddNTP is incorporated, initiating DNA synthesis until termination. The resulting DNA fragments are denatured into ssDNA.
Denatured fragments undergo gel electrophoresis for sequence determination. DNA polymerase synthesizes DNA only in the 5’ to 3’ direction, initiating at a provided primer. Each terminal ddNTP corresponds to a specific nucleotide in the original sequence. For example, the shortest fragment must terminate at the first nucleotide from the 5’ end, the second-shortest fragment must terminate at the second nucleotide from the 5’ end, and so on. Reading gel bands from smallest to largest reveals the 5’ to 3’ sequence of the original DNA strand.[41]
In manual Sanger sequencing, the user reads all 4 gel lanes simultaneously, moving from bottom to top to identify the terminal ddNTP for each band. For instance, if the bottom band is found in the ddGTP column, then the smallest PCR fragment terminates with ddGTP, and the first nucleotide from the 5’ end of the original sequence has a guanine (G) base.[42] Automated Sanger sequencing employs a computer to read each capillary gel band sequentially, using fluorescence to determine the terminal ddNTP identity. Laser activation of fluorescent tags emits light, detected by the computer, with each ddNTP tagged with a unique fluorescent label. The output is a chromatogram displaying fluorescent peaks corresponding to each nucleotide along the template DNA's length.[43]
Third-generation sequencing enables sequencing long DNA or RNA stretches without fragmentation. Single strands of DNA or RNA are directed through protein nanopores, with nucleotide bases distinguished by characteristic changes in electric current to determine the sequence.[44] Compared to 2nd-generation sequencing, 3rd-generation sequencing requires minimal sample preprocessing, enabling the design of smaller and more portable equipment.[45]
Indications
Molecular genetic testing has distinct indications, differing from traditional clinical and molecular biological testing used for diagnosing other diseases.[46] This modality’s applications encompass newborn screening, diagnostic testing for genetic or chromosomal conditions, carrier testing, prenatal testing, predictive and presymptomatic testing for adult-onset disorders, and forensic testing for legal identification purposes.[47]
FISH is employed for patients with a family history of known deletions and has been utilized to detect deletions in single blastomeres during preimplantation genetic diagnosis. FISH tests use gene-specific probe panels to investigate deletions, amplifications, and translocations in hematologic and solid tumors. FISH can also identify intracellular microorganisms and parasites.
CMA is recommended for individuals lacking specific clinical indicators to identify genetic or nongenetic causes of intellectual disability, developmental delay, autism spectrum disorder, or multiple congenital anomalies.[48] CMA can be helpful if prenatal structural anomalies are linked to particular microdeletions or microduplications. This modality can also evaluate copy number variants in cases of de novo balanced rearrangements or marker chromosomes.[49]
MLPA has diverse applications, such as mutation detection, single nucleotide polymorphisms (SNP) analysis, DNA methylation analysis, mRNA quantification, chromosomal characterization, gene copy number detection, and identification of duplications and deletions in cancer predisposition genes like BRCA1, BRCA2, hMLH1, and hMSH2. MLPA also holds promise for prenatal diagnosis, both invasive and noninvasive.[50]
DHPLC is well-suited for scanning genes for novel mutations and analyzing large sample sizes cost-effectively. This test is also useful for genotyping specific mutations or polymorphisms. DHPLC offers various applications beyond detecting genetic variants, including size-based double-strand DNA separation, single-strand DNA separation, and DNA purification analysis.[51]
NGS rapidly sequences whole genomes and target regions, employs RNA sequencing to identify novel RNA variants and splice sites, quantifies mRNAs for gene expression analysis, and analyzes epigenetic factors like DNA methylation and DNA-protein interactions. Sequence cancer samples study rare somatic variant tumor subclones and identify novel pathogens. Sanger sequencing, or the "chain termination method," determines DNA nucleotide sequences.
Potential Diagnosis
FISH swiftly diagnoses common fetal aneuploidies but with reduced sensitivity compared to cytogenetic analysis. FISH cannot identify cytogenetic abnormalities beyond the most common ones, such as translocations, inversions, and markers. DHPLC detects single nucleotide changes, small deletions, or insertions requiring subsequent confirmation by sequencing. This method identifies unknown mutations, making it advantageous for diseases with a high proportion of de novo mutations. Neurofibromatosis type 1 (NF1) is an example, as approximately 50% of cases arise from new mutations. CMAs are first-tier tests for developmental delays, intellectual disabilities, autism spectrum disorders, or multiple congenital disabilities, replacing karyotyping.
MLPA detects gene abnormalities, particularly small deletions in diseases like multiple endocrine neoplasia type 1 (MEN1 partial or complete deletion). MLPA can also assess methylation alterations, such as in pseudohypoparathyroidism 1b (PHP1b), where deletion of 1 or 4 of four differentially methylated regions is common.
NGS generates millions of sequences, which are then processed, analyzed, and interpreted to identify variants. Bioinformatics analysis begins with raw data generated by nucleotide incorporation signal detection. Read quality is evaluated during primary data analysis. Sequences are aligned or mapped against a reference genome, with computational algorithms searching for the best match for each read while allowing for some mismatches to detect genetic variants.[52]
Sanger sequencing is reliable in detecting point mutations, small deletions, or duplications. This method has a long history of use across various settings, including tumor mutational spectrum analysis and diagnostic testing for constitutional variants. Primers can cover multiple regions (amplicons) or any desired region size.
Normal and Critical Findings
The increasing demand for genetic testing has led to greater availability. Ensuring uniformity and standardization in communicating the complex results to referring clinicians is essential. Failure to include pertinent information is considered a deficiency in the molecular pathology laboratory accreditation inspection.[53] All molecular genetic laboratories offering clinical testing should be accredited according to the Clinical Laboratory Improvement Amendments and actively participate in proficiency testing.[54]
A comprehensive genetic report must include essential patient details such as name, medical record number or birth date, sex, and ethnicity. The report should also specify the type of specimen received, identification number, laboratory test requested, the performing laboratory’s name and address, and referring healthcare professional or hospital. The date of the report, analytic result interpretation using standard nomenclature, detailed method description (including literature citations if applicable), and assay sensitivity and specificity should be provided. For example, sensitivity and specificity should be reported regarding the number of variants analyzed, the proportion of variants not detected, and the possibility of genetic heterogeneity and recombination.[55] Reports from clinical DNA laboratories should include a disclaimer due to the prevalence of laboratory-developed tests (LDTs) or procedures (LDPs) designed, developed, and validated internally by each laboratory but remain unapproved by the FDA.[56]
Fluorescent tags binding to chromosomes reveal chromosomal abnormalities in FISH. MLPA detects copy number variations by correlating peak intensity during capillary electrophoresis with sample copy numbers. An MLPA probe's amplification signals the presence of a mutation in the sample.
An MLPA test can yield two outcomes:
- A positive result indicates the presence of a pathogenic or likely pathogenic gene deletion or duplication associated with the studied disease phenotype.
- A negative result suggests the absence of disease-causing deletions or duplications. However, this outcome does not guarantee overall health or the absence of other medical conditions or genetic disorders. Furthermore, a negative result does not exclude a genetic basis for the disease or eliminate risks for future offspring. In cases where affected family members carry the variant in question, a negative result in the proband excludes a diagnosis. Several factors, including limited genetic knowledge and methodological limitations, can produce a negative result.[57]
DHPLC detects mutations by identifying heteroduplexes compared to the reference genome in the same sample. NGS identifies various genetic variants, including single nucleotides, small insertions or deletions, and some structural variants, but their role in the disease is not implied. Clinical analysis and assessment of the pathological potential of detected variants require consideration in different contexts.[58] Sanger sequencing results interpretation depends on the target DNA strand and primer availability. If strand A is of interest but the primer suits strand B better, the output matches strand A. Conversely, if the primer suits strand A better, the output aligns with strand B, necessitating conversion back to strand A.
Interfering Factors
FISH probe specificity prevents unintended hybridization with nontarget genes. Some FISH preparations may exhibit autofluorescence, necessitating thorough cell washing to remove fluorescent residues and reduce background fluorescence.
MPLA has limitations, including its ability to detect only known mutations designed into probes, making gene rearrangements like inversions and translocations undetectable. Sample purity is essential as contaminants such as phenol can interfere with the ligation step. MLPA may yield false positive or negative results due to rare sequence variants in target regions detected by probes. Reduced probe binding efficiency from point mutations or polymorphisms can diminish the relative peak area’s height. Confirmation of single exon deletions detected by MLPA is thus recommended using other methods like multiplex PCR or sequencing.[59]
DHPLC sensitivity relies on melting temperature. Computational algorithms can predict the melting temperature, and the procedure typically involves at least 2 melting temperatures for increased sensitivity. CMA does not detect point mutations, small DNA segment changes (eg, in Fragile X syndrome), or balanced chromosomal rearrangements (eg, balanced translocations, inversions).
NGS technologies continue to evolve to address various challenges. Some large sequencers can detect large insertions, duplications, and deletions, while sequencing long homopolymer regions remains problematic. However, establishing the infrastructure and expertise for data analysis remains a significant challenge in clinical settings. The primary limitation of implementing NGS in clinical settings is the requirement for adequate infrastructure, including computational resources, storage capacity, and skilled personnel for comprehensive data analysis and interpretation.
Despite automation, Sanger sequencing remains labor-intensive, time-consuming, and expensive, relying on specialized equipment. Sanger sequencing exhibits reduced sensitivity in detecting point mutations when 20% of mutant DNA is of a wild-type background. Additionally, it lacks quantifiability, making it impossible to differentiate mutation prevalence accurately based solely on peak sizes, necessitating supplementary testing approaches.
Complications
Peripheral blood collection via venipuncture infrequently leads to serious complications. Some patients, especially children, may experience hematomas, pain, and fear, which are expected. In contrast, procedures like amniocentesis are more invasive, thus posing more serious risks such as infection, preterm delivery, respiratory distress, trauma, and alloimmunization, though these complications are also infrequent.[60] Genetic tests using NGS of free-cell DNA from maternal peripheral blood offer an alternative to diagnosis using amniocentesis fluid.[61]
Patient Safety and Education
Molecular testing may give rise to legal, medical, psychological, and ethical issues besides the sampling procedure’s potential complications.[62] While molecular testing primarily aims to demonstrate a genetic trait associated with a disease, the current recommendation is to integrate the results into genetic counseling.[63]
Genetic counseling, led by a team including genetic counselors and other professionals, begins with clinically identifying suspected diseases to guide molecular testing. Patients are informed about the testing procedure, potential results, and legal considerations like informed consent, particularly for children.[64] Patient education is integral to this process.
NGS technologies applied to genetic counseling yield complex results surpassing traditional tests, necessitating informed patient discussions due to the considerable information and ethical implications involved.[65] Laboratories conducting molecular genetic tests should address preexamination, examination, and postexamination considerations, tailoring methodology and interpretation to each test's indication, application, and ethical implications.
Clinical Significance
Any permanent alteration in a gene's nucleotide sequence compared to a reference genome is deemed a genetic change or mutation. Variants identified through a tiered protocol must undergo sequencing confirmation, and their role in disease pathology must be assessed. Genetic testing may reveal variants classified as benign, likely benign, pathogenic, likely pathogenic, or of uncertain significance.[66] Variants must be rigorously classified based on various types of evidence—population, computational, functional, or segregation data—to determine clinical significance.[67]
The American College of Medical Genetics and Genomics recommends this nomenclature and classification for genetic test findings, covering genotyping, single genes, panels, exomes, and genomes. NGS applications have deepened our understanding of genetic diseases and led to the discovery of variants requiring further study of their disease implications.[68] Interprofessional collaboration is essential for leveraging genetic tests for patient benefit, with an expert panel advocating for results interpretation by a board-certified geneticist.[69]
Molecular genetic testing advanced significantly with PCR and NGS, providing genome-wide data.[70] Multidisciplinary teams collaborate to integrate various testing methods with clinical, pathological, functional, computational, ethical, and social aspects of diseases for patient benefit.[71]