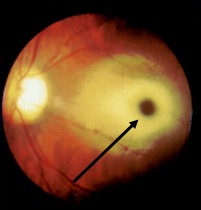
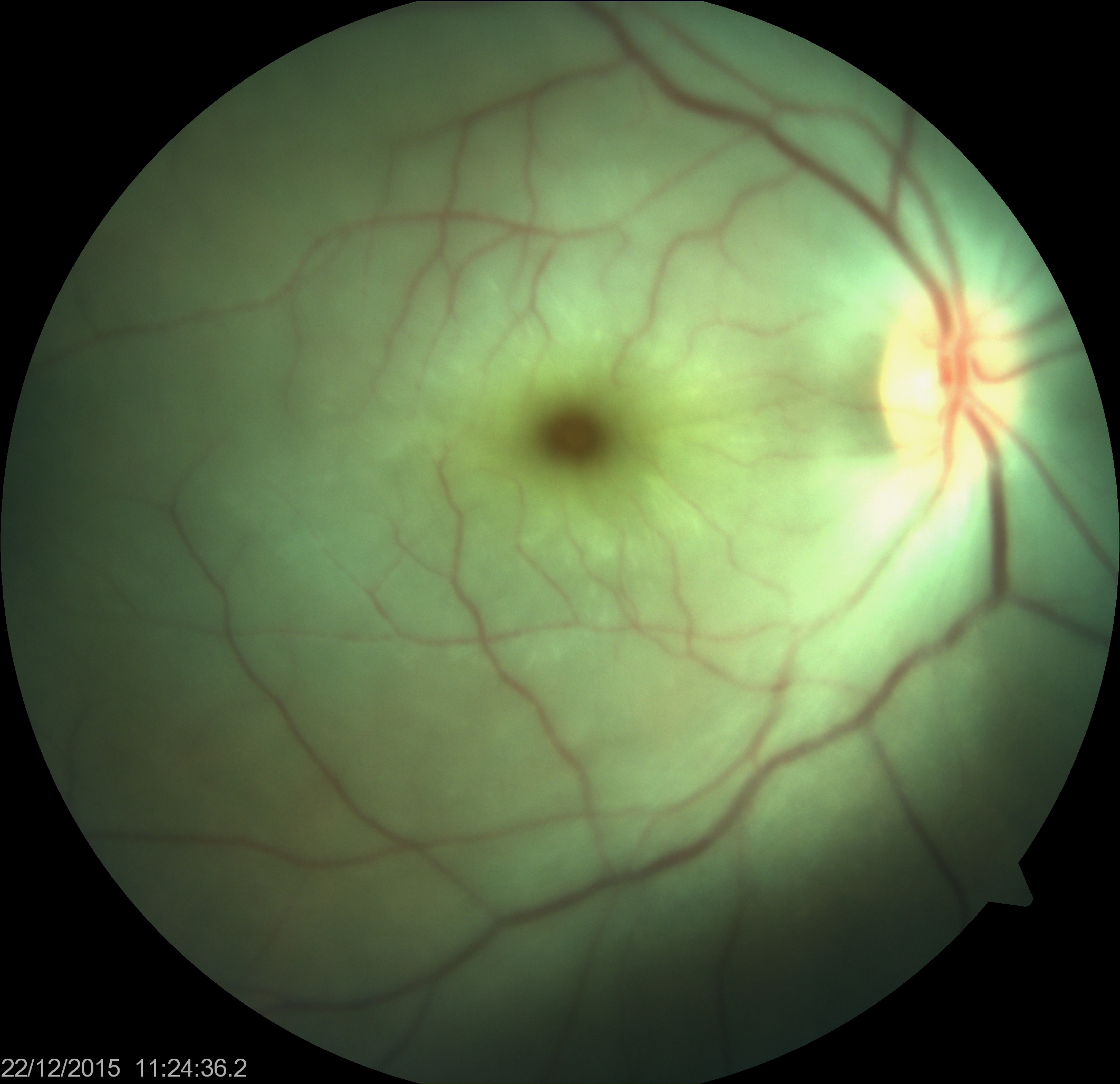
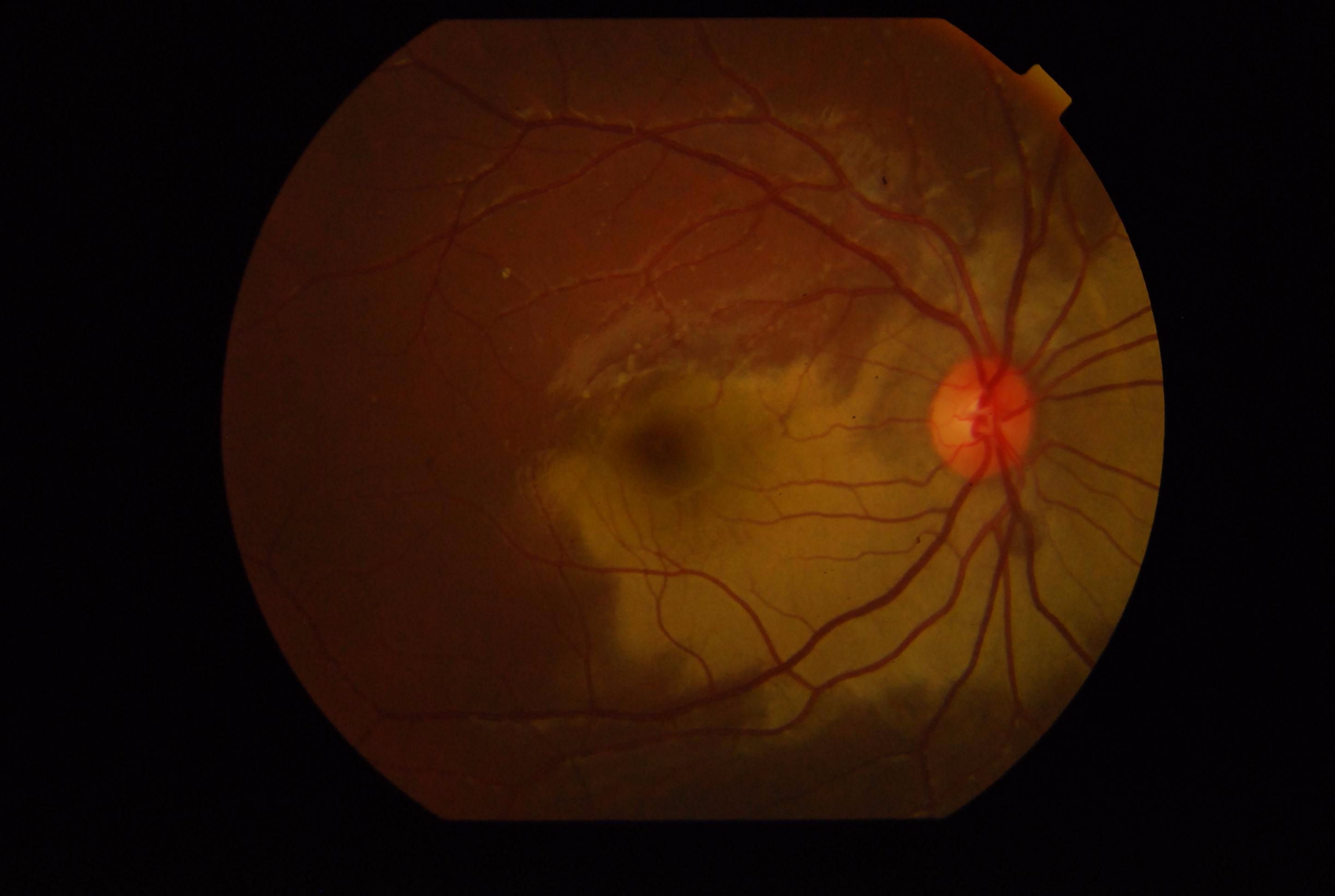
[1]
Hayreh SS, Acute retinal arterial occlusive disorders. Progress in retinal and eye research. 2011 Sep;
[PubMed PMID: 21620994]
[2]
Tripathy K, Mazumdar S, Sarma B. Central retinal arterial occlusion in a patient with pyoderma gangrenosum. Indian journal of ophthalmology. 2018 Jul:66(7):1019-1021. doi: 10.4103/ijo.IJO_1229_17. Epub
[PubMed PMID: 29941761]
[3]
Hayreh SS,Zimmerman MB, Fundus changes in central retinal artery occlusion. Retina (Philadelphia, Pa.). 2007 Mar;
[PubMed PMID: 17460582]
[4]
Jung EH,Park KH,Woo SJ, Iatrogenic Central Retinal Artery Occlusion Following Retrobulbar Anesthesia for Intraocular Surgery. Korean journal of ophthalmology : KJO. 2015 Aug
[PubMed PMID: 26240507]
[5]
Vasavada D,Baskaran P,Ramakrishnan S, Retinal Vascular Occlusion Secondary to Retrobulbar Injection: Case Report and Literature Review. Middle East African journal of ophthalmology. 2017 Jan-Mar
[PubMed PMID: 28546695]
Level 3 (low-level) evidence
[6]
Zhao C,Wei D,Shi X,Zhao M, Unilateral isolated optic nerve infiltration combined with central retinal artery occlusion in a patient with acute myeloid leukemia. American journal of ophthalmology case reports. 2022 Jun
[PubMed PMID: 35321253]
Level 3 (low-level) evidence
[7]
Tripathy K, Kumawat B, Chawla R, Sharma YR, Bypareddy R. Acute Vision Loss Due to Central Retinal Arterial Occlusion, Partial Optic Nerve Avulsion, and Hemorrhage "Spurting Out" from Optic Disc after Blunt Trauma. Journal of ophthalmic & vision research. 2017 Jul-Sep:12(3):351-352. doi: 10.4103/jovr.jovr_4_15. Epub
[PubMed PMID: 28791073]
[8]
Goel N,Rajput M,Sawhney A,Sardana T, Macular infarction and traumatic optic neuropathy following blunt ocular trauma. Saudi journal of ophthalmology : official journal of the Saudi Ophthalmological Society. 2016 Jan-Mar;
[PubMed PMID: 26949360]
[9]
Ahmed NR,Tripathy K,Kumar V,Gogia V, Choroidal coloboma in a case of tay-sachs disease. Case reports in ophthalmological medicine. 2014
[PubMed PMID: 25295204]
Level 3 (low-level) evidence
[10]
Tripathy K,Chawla R, Extensive commotio retinae involving peripheral retina. The National medical journal of India. 2017 Jul-Aug;
[PubMed PMID: 29162764]
[11]
Chen H,Chan AY,Stone DU,Mandal NA, Beyond the cherry-red spot: Ocular manifestations of sphingolipid-mediated neurodegenerative and inflammatory disorders. Survey of ophthalmology. 2014 Jan-Feb;
[PubMed PMID: 24011710]
Level 3 (low-level) evidence
[13]
Tripathy K,Chawla R,Mittal K,Farmania R,Venkatesh P,Gulati S, Ophthalmic examination as a means to diagnose Subacute Sclerosing Panencephalitis: an optical coherence tomography and ultrawide field imaging evaluation. Eye and vision (London, England). 2017
[PubMed PMID: 28116334]
[14]
Chawla R,Tripathy K,Gogia V,Venkatesh P, Progressive outer retinal necrosis-like retinitis in immunocompetent hosts. BMJ case reports. 2016 Aug 10;
[PubMed PMID: 27511757]
Level 3 (low-level) evidence
[15]
Yiu G,Young LH, Progressive outer retinal necrosis presenting as cherry red spot. Ocular immunology and inflammation. 2012 Oct;
[PubMed PMID: 22957726]
[16]
Abhayambika K,Chacko A,Mahadevan K,Najeeb OM, Peripheral neuropathy and haemolytic anaemia with cherry red spot on macula in dapsone poisoning. The Journal of the Association of Physicians of India. 1990 Aug;
[PubMed PMID: 2174032]
[17]
Goker-Alpan O,Schiffmann R,Park JK,Stubblefield BK,Tayebi N,Sidransky E, Phenotypic continuum in neuronopathic Gaucher disease: an intermediate phenotype between type 2 and type 3. The Journal of pediatrics. 2003 Aug
[PubMed PMID: 12970647]
[18]
Winter AW,Salimi A,Ospina LH,Roos JCP, Ophthalmic manifestations of Gaucher disease: the most common lysosomal storage disorder. The British journal of ophthalmology. 2019 Mar
[PubMed PMID: 30612093]
[19]
Lew RM,Burnett L,Proos AL,Delatycki MB, Tay-Sachs disease: current perspectives from Australia. The application of clinical genetics. 2015;
[PubMed PMID: 25653550]
Level 3 (low-level) evidence
[20]
Rumelt S, Dorenboim Y, Rehany U. Aggressive systematic treatment for central retinal artery occlusion. American journal of ophthalmology. 1999 Dec:128(6):733-8
[PubMed PMID: 10612510]
Level 1 (high-level) evidence
[23]
Ferreira CR, Gahl WA. Lysosomal storage diseases. Translational science of rare diseases. 2017 May 25:2(1-2):1-71. doi: 10.3233/TRD-160005. Epub 2017 May 25
[PubMed PMID: 29152458]
[24]
Hayreh SS,Zimmerman MB,Kimura A,Sanon A, Central retinal artery occlusion. Retinal survival time. Experimental eye research. 2004 Mar
[PubMed PMID: 15106952]
[31]
Heroman JW,Rychwalski P,Barr CC, Cherry red spot in sialidosis (mucolipidosis type I). Archives of ophthalmology (Chicago, Ill. : 1960). 2008 Feb;
[PubMed PMID: 18268224]
[32]
Wang IH,Lin TY,Kao ST, Optical coherence tomography features in a case of Type I sialidosis. Taiwan journal of ophthalmology. 2017 Apr-Jun
[PubMed PMID: 29018767]
Level 3 (low-level) evidence
[33]
Ahn SJ,Woo SJ,Kim KE,Jo DH,Ahn J,Park KH, Optical coherence tomography morphologic grading of macular commotio retinae and its association with anatomic and visual outcomes. American journal of ophthalmology. 2013 Nov;
[PubMed PMID: 23972302]
[34]
Zou W,Wang X,Tian G, Fundus autofluorescence and optical coherence tomography of a macular cherry-red spot in a case report of sialidosis. BMC ophthalmology. 2016 Mar 22;
[PubMed PMID: 27004518]
Level 3 (low-level) evidence
[35]
Mathew R,Papavasileiou E,Sivaprasad S, Autofluorescence and high-definition optical coherence tomography of retinal artery occlusions. Clinical ophthalmology (Auckland, N.Z.). 2010 Oct 21;
[PubMed PMID: 21060665]
[36]
Varma DD,Cugati S,Lee AW,Chen CS, A review of central retinal artery occlusion: clinical presentation and management. Eye (London, England). 2013 Jun
[PubMed PMID: 23470793]
[37]
Tripathy K,Venkatesh P,Chawla R, Cytomegaloviral retinitis. The National medical journal of India. 2016 Jan-Feb
[PubMed PMID: 27492038]
[38]
Chawla R,Tripathy K,Temkar S,Venkatesh P,Kumar A, An imaging-based treatment algorithm for posterior focal retinitis. Therapeutic advances in ophthalmology. 2018 Jan-Dec
[PubMed PMID: 29998221]
Level 3 (low-level) evidence
[39]
Park JY,Nam WH,Kim SH,Jang SY,Ohn YH,Park TK, Evaluation of the central macula in commotio retinae not associated with other types of traumatic retinopathy. Korean journal of ophthalmology : KJO. 2011 Aug
[PubMed PMID: 21860574]