Continuing Education Activity
Congenital hypertrophy of the retinal pigmented epithelium (CHRPE) is a commonly encountered, generally benign, non-progressive lesion within the retinal pigmented epithelial (RPE) cell layer of the outer retina. RPE cells are normally responsible for providing functional support to the inner (neural) retina and are crucial in maintaining metabolic homeostasis. Histological analysis shows changes in the cell size, cell quantity, and the number of melanin (pigment) granules within affected RPE cells. The name "CHRPE" is often used clinically to describe other retinal lesions that are similar in appearance but possess vital differences in prognosis. This activity outlines the evaluation, differentiation, and management of CHRPE lesions and reviews the role of the interprofessional team in evaluating and managing patients with this condition.
Objectives:
- Identify the funduscopic appearance of CHRPE lesions and how they differ from the appearance of PO-FLs.
- Describe the histological characteristics of RPE cells within solitary CHRPE lesions and contrast with GPR and PO-FLs.
- Review the typical presentation of solitary CHRPE lesions when evaluated with ocular coherence tomography (OCT) and fluorescein angiography (FA).
- Outline the differences in prognosis, proper management strategy, and screening considerations for patients presenting with solitary CHRPE, GPR, and PO-FLs.
Introduction
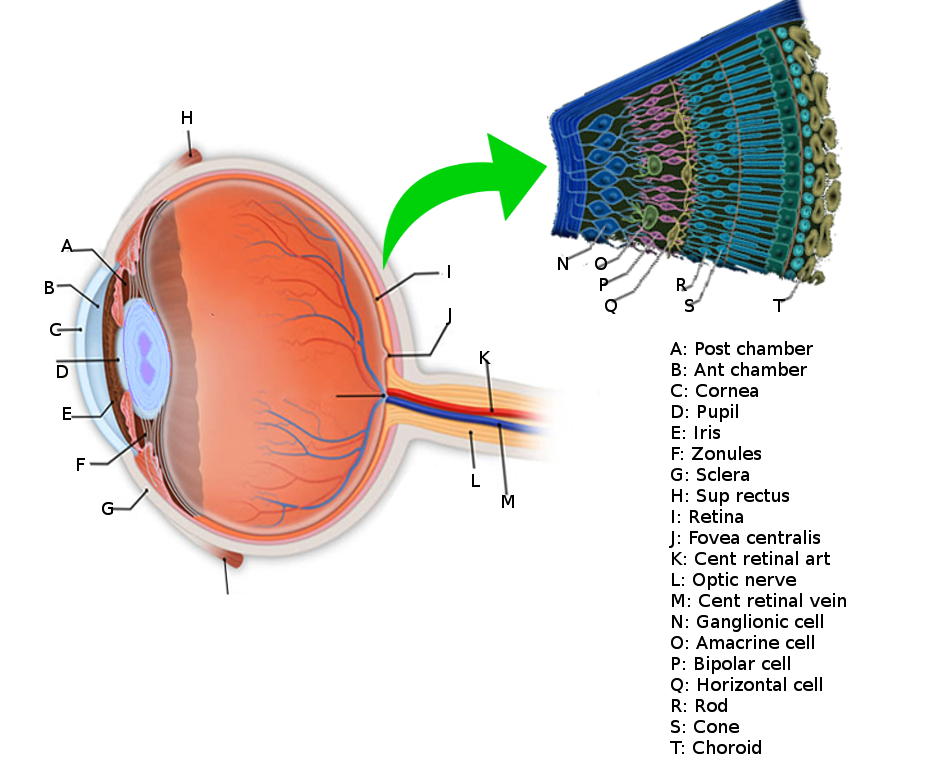
The retina is an ocular structure responsible for gathering gross visual stimuli that enter the eye and transforming them into neuronal signals transported to the brain for interpretation. Truly, it comprises two distinct entities that work in concert with each other: the neural retina and the retinal pigmented epithelium. The underlying choroid and its blood supply also play a crucial role in this process; these three structures must work together to maintain homeostatic balance and function properly.[1] Disruption of the homeostatic balance between the RPE and neural retina is the basis for many of the most common retinal diseases.
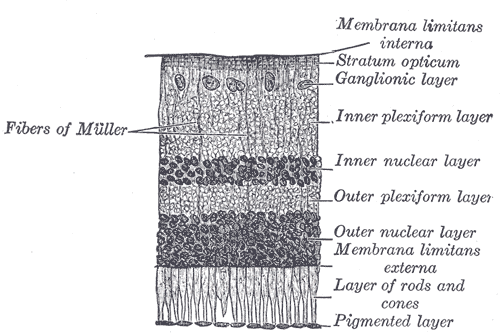
The Neural Retina
The photoreceptor layer houses the cell bodies of rod and cone photoreceptors; these cells contain visual pigments responsible for absorbing light and triggering the conversion of light photons into neurochemical signals, a process known as phototransduction. The neural retina comprises seven layers of various photosensitive neurons that synapse on one another along a cascade that ultimately terminates in the visual cortex of the occipital lobe for interpretation.[2]
The Retinal Pigmented Epithelium
Outer to the neural retina is the retinal pigmented epithelium (RPE), a single (simple) layer of cuboidal epithelial cells joined together by zonula occludens junctions. These tight junctions allow RPE cells to act as a gate, containing bidirectional pumps that regulate the transport of molecules to and from the choroid and neural retina.[3] Another notable feature of RPE cells is their high concentration of intracellular melanosomes, a cellular organelle that produces pigment granules known as melanin. These molecules act to absorb stray light and neutralize free radical oxygen species.[4]
These unique characteristics allow the RPE to provide functional support to the neural retina and act to maintain metabolic homeostasis via:
- Digestion of photoreceptor outer segments and recycling of used photopigments during the visual cycle
- Maintenance of the outer blood-retinal barrier via intercellular tight junctions
- Reduction of oxidative stress
The Choroid
The underlying choroid is a vascular structure that supplies oxygen and nutrients to the RPE and the majority (approximately 85%) of the retina. Its large diameter, fenestrated vessels are specialized to support the extensive metabolic demand of the photoreceptors and RPE.
This article focuses on certain histologic changes in RPE cells and disruption of retinal homeostasis. The changes described here are the basis for developing retinal lesions (and associated lesions) known as congenital hypertrophy of the RPE.[5]
Etiology
Congenital hypertrophy of the retinal pigmented epithelium (CHRPE) is a term that is frequently used to describe a group of conditions that share many clinical features but differ significantly in etiology, histopathology, prognosis, and management. The following names and abbreviations have been gathered from various studies to more accurately describe and differentiate between these conditions.
- Typical CHRPEs are generally unilateral and solitary lesions that are not associated with systemic conditions. They develop sporadically and are almost always benign. These are the only 'true' CHRPEs.[6]
- Grouped pigmentation of the retina (GPR) is also unilateral, and lesions appear similar to CHRPE but are multifocal and present in clusters within a single quadrant of the retina. They are commonly referred to as "bear tracks" and are also congenital and benign in nature, with no associated systemic conditions.[7]
- Pigmented ocular fundus lesions of familial adenomatous polyposis (PO-FLs) are multifocal, similar to GPR; however, lesions are typically bilateral. Unlike other CHRPE variants, PO-FLs are associated with a group of hereditary genetic colonic polyposis conditions, described below.
Familial Adenomatous Polyposis (FAP) is an autosomal dominant inherited condition that results from a mutation in the APC tumor suppressor gene. This condition is characterized by the development of hundreds of polyps within the colon that progresses to colorectal cancer in 95% of patients if left untreated. It often presents with extracolonic manifestations, one of which being CHRPE-appearing lesions in the retina.[7] Gardner syndrome and Turcot's syndrome are variants of FAP that are also associated with the development of PO-FLs. Due to the potentially dire prognosis of this condition, it is crucial for the eye care professional to be able to distinguish PO-FLs from their benign counterparts.[8]
Epidemiology
Prevalence rates of typical CHRPE have not been well defined in the literature and show significant variability between studies (0.4 to 30%).[9] Coleman et al. found a prevalence rate of 1.20% amongst a group of 1,745 study subjects. Only 2 of these subjects presented with multifocal lesions (prevalence <1%), and none presented bilaterally. Numerous studies have found no sexual predilection for typical CHRPE lesions.[6]
Mirinezhad et al. found that the presence of multiple pigmented ocular fundus lesions was compassionate (77%) and specific (60%) as clinical markers for FAP. There was no predilection for age, gender, and the number of lesions.[10]
Pathophysiology
Solitary and multifocal CHRPEs are sporadic, benign abnormalities of RPE cells, also known as retinal hamartomas. Histopathologic changes in RPE cell size, number, and content result in cellular malfunction and loss of homeostasis between the RPE, choroid, and neural retina of the affected area.[5]
Histopathology
Although the term "CHRPE" exclusively suggests hypertrophic cellular changes, histological analysis shows various morphologic cellular changes of CHRPE and its variants.
RPE cells in typical CHRPEs exhibit various degrees of cellular hypertrophy (increase in cell size) – normal, cuboidal RPE cells are vertically elongated and columnar in shape. Multiple studies, however, have found that cells also undergo hyperplasia (increase in cell number), resulting in thickening of the RPE layer. These changes disrupt the ability of the RPE to support the overlying photoreceptors, resulting in cell loss and thinning. Without photoreceptors, there is no production of lipofuscin - waste products secondary to the normal RPE-mediated phagocytosis of photoreceptor outer segments.[7]
Areas of atrophy, known as lacunae, may also occur throughout the lesion and correlate with depigmented funduscopic appearance. Compared to normal cuboidal RPE cells, affected RPE cells are vertically elongated and range from cuboidal to columnar in shape. They possess large, spherical intracytoplasmic melanin granules in much higher density than normal RPE cells. The adjacent Bruch's membrane is thickened.[11]
As opposed to its solitary counterpart, GPR has been found to contain histologically normal cells, with no difference in cell size or density compared to normal RPE cells. Instead, its hyperpigmented appearance results from the increased density of melanin granules, which are large and ellipsoid in shape. Bruch's membrane is normal in these cases.
Lastly, PO-FLs have been shown to be histologically similar to typical lesions, with both hypertrophic and hyperplastic RPE cells and thickening of the adjacent Bruch's membrane.[6][7]
History and Physical
Typical CHRPE
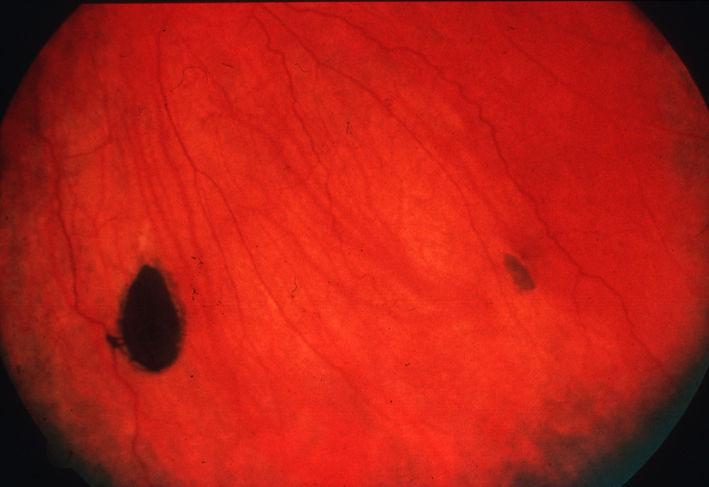
Lesions are flat (or minimally elevated) and highly pigmented, appearing black in color. In some cases, lesions may lack pigment entirely (12%) - these are referred to as amelanotic lesions. Lesion borders are well-defined and are smooth or scalloped in appearance. In 52% of patients, there is a ring of hypopigmentation or "halo" surrounding the borders of the lesion. Solitary CHRPEs are most commonly found in the mid- or far-peripheral fundus of the temporal quadrant of the retina. In rare cases (2%), lesions may be peripapillary or macular in location.[12]
Approximately half (43%) of pigmented CHRPEs present with hypopigmented areas of RPE atrophy, known as lacunae, within the lesion. On average, about 56.5% of the lesion surface is atrophic; however, the presence, appearance, and extent of the atrophy vary between patients and do not change the patient's prognosis.
The mean size of solitary CHRPEs is estimated to be 4.67 +/- 1.21 mm^2. Although the lesion is generally regarded as non-progressive and stationary, numerous studies have demonstrated slow, benign enlargement over time in up to 83% of patients.[13] In addition, 32% of cases also show enlargement of lacunae over time.
Lesions are most often found incidentally during a routine eye examination, and patients are rarely symptomatic. Visual field defects corresponding to the area of the lesion are possible; however, lesions are generally located peripherally and are unlikely to be noticeable.
Grouped Pigmentation of the Retina (GPR)
Grouped pigmentation of the retina, often called "bear tracks," appears similarly to solitary CHRPE but presents unilaterally with anywhere from 3-30 discreet lesions of various sizes. They are generally smaller than unilateral cases, ranging from 0.1 to 0.3 mm in diameter, and confined to a single quadrant within the retina. These lesions differ from solitary CHRPE in that they generally lack surrounding haloes and lacunae.[5]
Pigmented Ocular Fundus Lesions of Familial Adenomatous Polyposis (PO-FLs)
Due to their association with serious systemic disease, clinicians must accurately differentiate between PO-FLs and benign CHRPE. As opposed to their benign counterpart, PO-FLs are bilateral and multifocal, with lesions in more than one quadrant. They often have a "pisciform" (fish-shaped) appearance and irregular, depigmented borders.[7]
Evaluation
Dilated Fundus Examination
As mentioned, CHRPE lesions are rarely found in the posterior pole. Therefore, most cases cannot be detected without evaluating the retinal periphery via dilated examination or non-mydriatic retinal imaging. One or both procedures are virtually always performed as part of a comprehensive eye exam. Most biomicroscopes are equipped with a red-free filter that may be useful in differentiating between choroidal nevi and CHRPE lesions. Nevi will disappear with red-free, while CHRPE will remain visible.[14]
Optical Coherence Tomography (OCT)
OCT shows hyperreflectivity and thickening of the RPE corresponding to the abnormally elongated RPE cells. Thickening of the RPE results in underlying choroidal shadowing. There is retinal thinning overlying the lesion and loss of the photoreceptor layer.[15]
A study by Francis et al. found evidence of associated choroidal atrophy when lesions were imaged with SD-OCT. They found that the inner layers of the choroid, i.e., the choriocapillaris and Sattler's layer, were atrophied in 97% of their cohort. Larger diameter lesions were more likely to involve the outer choroid, i.e., Haller's layer; 53% of patients showed outer choroidal involvement.[16]
Fluorescein Angiography (FA)
FA reveals hypofluorescence of lesions secondary to the "block effect" - this term refers to the signal blockage due to the hypertrophic RPE cells anterior to the choroidal vasculature preventing visualization of normal background fluorescein leakage. If present, lacunae within the lesion will appear hyperfluorescent.[17] In some cases, changes in retinal vasculature may occur, which can be detected with FA, including capillary nonperfusion areas, capillary microaneurysms, dye leakage, and chorioretinal anastomosis.
Fundus Autofluorescence (FAF)
Lesions appear strongly hypoautofluorescent when imaged with FAF; however, lacunae (if present) appear slightly hyperautofluorescent.[18]
B-Scan Ultrasonography
B-scan is not diagnostic for CHRPE but may be used to determine the thickness of the lesion. It is most commonly 0.5-1.0 mm thick.[5]
Visual Field Analysis (VFA)
Although it is possible for patients to manifest a visual field defect corresponding to the area of the CHRPE, they are unlikely to be detected with VFA. Most lesions are more peripheral than the testing area of Humphrey visual field analyzers. Depth of the defect (absolute vs. relative) is generally related to the size of the lesion; however, some studies suggest that younger patients generally manifest relative visual field defects, while older patients manifest absolute defects.[15]
Treatment / Management
Solitary CHRPE and GPR do not require treatment and may be observed periodically. Solitary lesions should be closely examined for the rare instance of nodule development and associated complications or malignant transformation (see section "Prognosis"). The lesion may be measured, and fundus photography may be employed to monitor for changes in lesion size/characteristics.
Patients with PO-FLs should undergo routine endoscopies with their gastroenterologist to assess for the presence of polyps within the colon and rectum. Genetic screening should be performed in patients with a family history of adenomatous polyposis syndrome or patients with ten or more polyps regardless of family history.[19] Genetic determination of the specific mutation dictates the recommended frequency of screening and prognosis. The presence of solitary or multifocal CHRPEs is not associated with an increased risk of systemic disease and does not warrant further testing or referral.[7]
Differential Diagnosis
Differential diagnoses of CHRPE include:
- Choroidal nevus
- Choroidal melanoma
- Melanocytoma of the optic nerve head
- Congenital simple RPE hamartoma
- Combined hamartoma of the retina and RPE
- Acquired RPE hyperplasia[6]
Prognosis
Virtually all patients with solitary CHRPE or GPR are asymptomatic, and lesions are benign; they are not associated with an increased risk of systemic disease. The prognosis is very good.
Although rare (2%), there is evidence of benign (adenoma) or malignant (adenocarcinoma) transformation of RPE cells within solitary CHRPE lesions. They initially arise from the CHRPE as a small nodule that grows slowly, eventually invading the sensory retina and spawning its own vasculature. Feeder and drainage vessels carry lipoproteinaceous material to and from the lesion. The nodules may remain stable or lead to extensive intraocular inflammation, subretinal exudation, retinal detachment, and subsequent severe vision loss. Other complications include vitreous traction, cataract, and posterior synechiae. Uncomplicated cases are often monitored, as there is no reported incidence of metastasis.[13]
Complications
Complications associated with CHRPE are exceedingly rare and are generally secondary to nodular growth within the lesion. Subretinal fluid or exudation from nodules may occur, causing retinal detachment: a separation of the RPE from the neural retina. In addition, nodules may cause secondary vitreomacular traction and decreased central vision.[5]
Deterrence and Patient Education
Patients should be educated regarding the diagnosis of CHRPE, and emphasis should be placed on the importance of regular eye examinations. The patient should be educated regarding the possibility of transformation, with reassurance that malignancy is exceeding rare and unlikely.[5]
Upon initial presentation of suspected PO-FLs, patients should be educated thoroughly regarding the association with systemic polyposis syndromes and the development of colon cancer. The patient should be instructed to undergo further evaluation by a gastroenterologist.[7]
Enhancing Healthcare Team Outcomes
Solitary CHRPE and GPR are most likely to be identified by an eye care professional, such as an optometrist or ophthalmologist. Especially in suspicious cases where a second opinion is needed, the two professions work together in patient diagnosis and management.
In addition, PO-FLs require interprofessional management between the patient's eye care professional and gastroenterologist. It is the responsibility of the eye care professional to ensure that the patient receives an appropriate, timely referral and evaluation to improve the patient's outcome best.