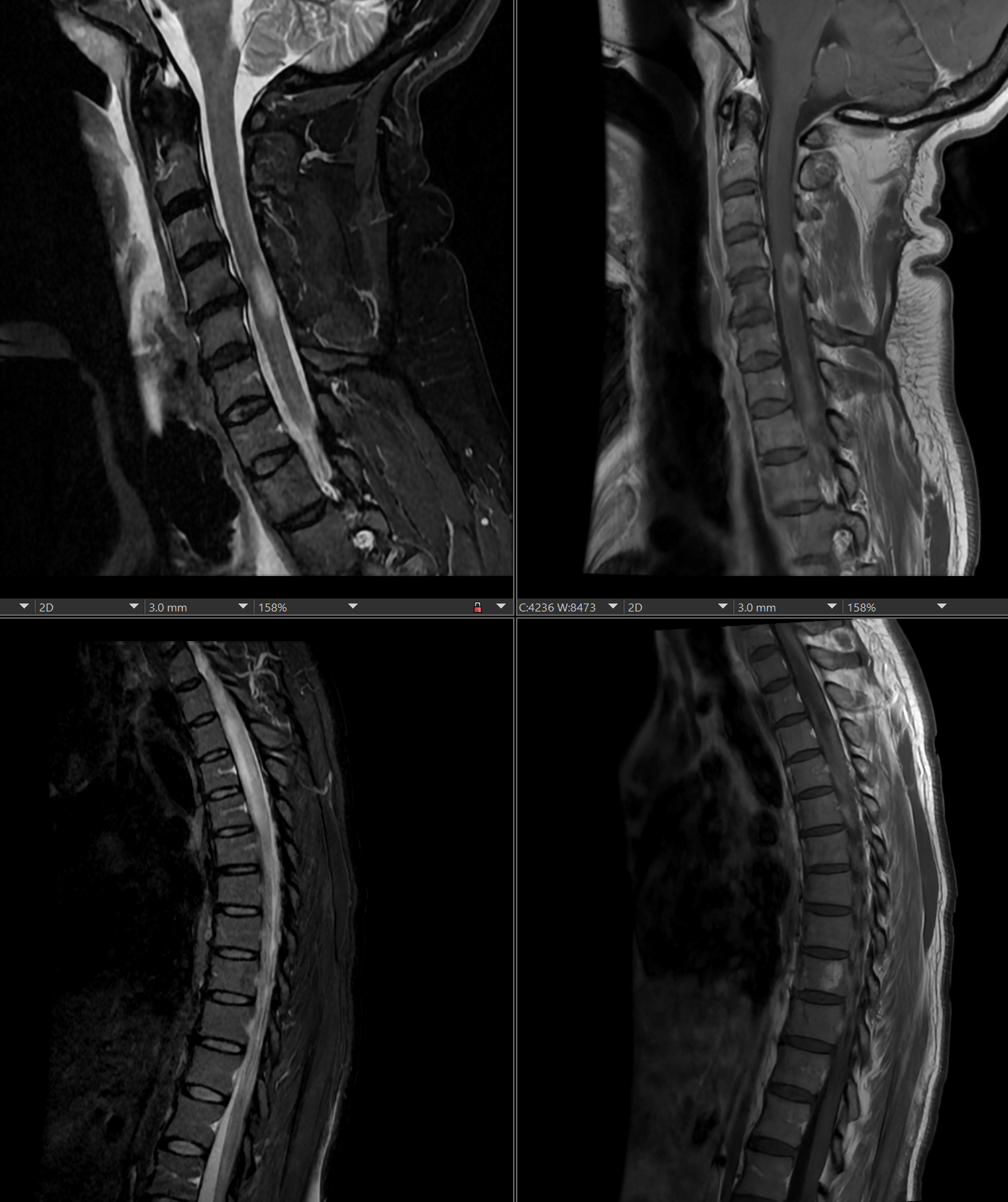
[1]
Jarius S, Wildemann B. The history of neuromyelitis optica. Journal of neuroinflammation. 2013 Jan 15:10():8. doi: 10.1186/1742-2094-10-8. Epub 2013 Jan 15
[PubMed PMID: 23320783]
[3]
Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T, de Seze J, Fujihara K, Greenberg B, Jacob A, Jarius S, Lana-Peixoto M, Levy M, Simon JH, Tenembaum S, Traboulsee AL, Waters P, Wellik KE, Weinshenker BG, International Panel for NMO Diagnosis. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015 Jul 14:85(2):177-89. doi: 10.1212/WNL.0000000000001729. Epub 2015 Jun 19
[PubMed PMID: 26092914]
Level 3 (low-level) evidence
[4]
Estrada K, Whelan CW, Zhao F, Bronson P, Handsaker RE, Sun C, Carulli JP, Harris T, Ransohoff RM, McCarroll SA, Day-Williams AG, Greenberg BM, MacArthur DG. A whole-genome sequence study identifies genetic risk factors for neuromyelitis optica. Nature communications. 2018 May 16:9(1):1929. doi: 10.1038/s41467-018-04332-3. Epub 2018 May 16
[PubMed PMID: 29769526]
[5]
Papadopoulos MC, Verkman AS. Aquaporin 4 and neuromyelitis optica. The Lancet. Neurology. 2012 Jun:11(6):535-44. doi: 10.1016/S1474-4422(12)70133-3. Epub 2012 May 16
[PubMed PMID: 22608667]
[6]
Ransohoff RM. Illuminating neuromyelitis optica pathogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2012 Jan 24:109(4):1001-2. doi: 10.1073/pnas.1119288109. Epub 2012 Jan 23
[PubMed PMID: 22308524]
[7]
Kitley J, Leite MI, Nakashima I, Waters P, McNeillis B, Brown R, Takai Y, Takahashi T, Misu T, Elsone L, Woodhall M, George J, Boggild M, Vincent A, Jacob A, Fujihara K, Palace J. Prognostic factors and disease course in aquaporin-4 antibody-positive patients with neuromyelitis optica spectrum disorder from the United Kingdom and Japan. Brain : a journal of neurology. 2012 Jun:135(Pt 6):1834-49. doi: 10.1093/brain/aws109. Epub 2012 May 9
[PubMed PMID: 22577216]
[8]
Pandit L, Asgari N, Apiwattanakul M, Palace J, Paul F, Leite MI, Kleiter I, Chitnis T, GJCF International Clinical Consortium & Biorepository for Neuromyelitis Optica. Demographic and clinical features of neuromyelitis optica: A review. Multiple sclerosis (Houndmills, Basingstoke, England). 2015 Jun:21(7):845-53. doi: 10.1177/1352458515572406. Epub 2015 Apr 28
[PubMed PMID: 25921037]
[9]
Cabrera-Gómez JA, Kurtzke JF, González-Quevedo A, Lara-Rodríguez R. An epidemiological study of neuromyelitis optica in Cuba. Journal of neurology. 2009 Jan:256(1):35-44. doi: 10.1007/s00415-009-0009-0. Epub 2009 Feb 9
[PubMed PMID: 19224310]
Level 2 (mid-level) evidence
[10]
Kim SH, Mealy MA, Levy M, Schmidt F, Ruprecht K, Paul F, Ringelstein M, Aktas O, Hartung HP, Asgari N, Tsz-Ching JL, Siritho S, Prayoonwiwat N, Shin HJ, Hyun JW, Han M, Leite MI, Palace J, Kim HJ. Racial differences in neuromyelitis optica spectrum disorder. Neurology. 2018 Nov 27:91(22):e2089-e2099. doi: 10.1212/WNL.0000000000006574. Epub 2018 Oct 26
[PubMed PMID: 30366977]
[11]
Trebst C, Jarius S, Berthele A, Paul F, Schippling S, Wildemann B, Borisow N, Kleiter I, Aktas O, Kümpfel T, Neuromyelitis Optica Study Group (NEMOS). Update on the diagnosis and treatment of neuromyelitis optica: recommendations of the Neuromyelitis Optica Study Group (NEMOS). Journal of neurology. 2014 Jan:261(1):1-16. doi: 10.1007/s00415-013-7169-7. Epub 2013 Nov 23
[PubMed PMID: 24272588]
[12]
Jarius S, Ruprecht K, Wildemann B, Kuempfel T, Ringelstein M, Geis C, Kleiter I, Kleinschnitz C, Berthele A, Brettschneider J, Hellwig K, Hemmer B, Linker RA, Lauda F, Mayer CA, Tumani H, Melms A, Trebst C, Stangel M, Marziniak M, Hoffmann F, Schippling S, Faiss JH, Neuhaus O, Ettrich B, Zentner C, Guthke K, Hofstadt-van Oy U, Reuss R, Pellkofer H, Ziemann U, Kern P, Wandinger KP, Bergh FT, Boettcher T, Langel S, Liebetrau M, Rommer PS, Niehaus S, Münch C, Winkelmann A, Zettl U UK, Metz I, Veauthier C, Sieb JP, Wilke C, Hartung HP, Aktas O, Paul F. Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: A multicentre study of 175 patients. Journal of neuroinflammation. 2012 Jan 19:9():14. doi: 10.1186/1742-2094-9-14. Epub 2012 Jan 19
[PubMed PMID: 22260418]
[13]
Wingerchuk DM, Hogancamp WF, O'Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic's syndrome). Neurology. 1999 Sep 22:53(5):1107-14
[PubMed PMID: 10496275]
[14]
Okada K, Kobata M, Naruke S. Neuromyelitis optica spectrum disorder with area postrema syndrome. Neurology. Clinical practice. 2019 Apr:9(2):173-175. doi: 10.1212/CPJ.0000000000000586. Epub
[PubMed PMID: 31041136]
[15]
Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. The Journal of experimental medicine. 2005 Aug 15:202(4):473-7
[PubMed PMID: 16087714]
[16]
Weinshenker BG, Wingerchuk DM. Neuromyelitis Spectrum Disorders. Mayo Clinic proceedings. 2017 Apr:92(4):663-679. doi: 10.1016/j.mayocp.2016.12.014. Epub
[PubMed PMID: 28385199]
[17]
Barnett Y, Sutton IJ, Ghadiri M, Masters L, Zivadinov R, Barnett MH. Conventional and advanced imaging in neuromyelitis optica. AJNR. American journal of neuroradiology. 2014 Aug:35(8):1458-66. doi: 10.3174/ajnr.A3592. Epub 2013 Jun 13
[PubMed PMID: 23764723]
[18]
Sarbu N, Shih RY, Jones RV, Horkayne-Szakaly I, Oleaga L, Smirniotopoulos JG. White Matter Diseases with Radiologic-Pathologic Correlation. Radiographics : a review publication of the Radiological Society of North America, Inc. 2016 Sep-Oct:36(5):1426-47. doi: 10.1148/rg.2016160031. Epub
[PubMed PMID: 27618323]
[19]
Romeo AR, Segal BM. Treatment of neuromyelitis optica spectrum disorders. Current opinion in rheumatology. 2019 May:31(3):250-255. doi: 10.1097/BOR.0000000000000603. Epub
[PubMed PMID: 30920972]
Level 3 (low-level) evidence
[20]
Wingerchuk DM. Evidence for humoral autoimmunity in neuromyelitis optica. Neurological research. 2006 Apr:28(3):348-53
[PubMed PMID: 16687064]
[21]
Gold R, Linington C. Devic's disease: bridging the gap between laboratory and clinic. Brain : a journal of neurology. 2002 Jul:125(Pt 7):1425-7
[PubMed PMID: 12076994]
[23]
Patterson SL, Goglin SE. Neuromyelitis Optica. Rheumatic diseases clinics of North America. 2017 Nov:43(4):579-591. doi: 10.1016/j.rdc.2017.06.007. Epub 2017 Aug 31
[PubMed PMID: 29061244]
[25]
de Seze J, Blanc F, Jeanjean L, Zéphir H, Labauge P, Bouyon M, Ballonzoli L, Castelnovo G, Fleury M, Defoort S, Vermersch P, Speeg C. Optical coherence tomography in neuromyelitis optica. Archives of neurology. 2008 Jul:65(7):920-3. doi: 10.1001/archneur.65.7.920. Epub
[PubMed PMID: 18625858]
[26]
Nakamura M, Misu T, Fujihara K, Miyazawa I, Nakashima I, Takahashi T, Watanabe S, Itoyama Y. Occurrence of acute large and edematous callosal lesions in neuromyelitis optica. Multiple sclerosis (Houndmills, Basingstoke, England). 2009 Jun:15(6):695-700. doi: 10.1177/1352458509103301. Epub 2009 May 12
[PubMed PMID: 19435750]
[27]
Kim HJ, Paul F, Lana-Peixoto MA, Tenembaum S, Asgari N, Palace J, Klawiter EC, Sato DK, de Seze J, Wuerfel J, Banwell BL, Villoslada P, Saiz A, Fujihara K, Kim SH, Guthy-Jackson Charitable Foundation NMO International Clinical Consortium & Biorepository. MRI characteristics of neuromyelitis optica spectrum disorder: an international update. Neurology. 2015 Mar 17:84(11):1165-73. doi: 10.1212/WNL.0000000000001367. Epub 2015 Feb 18
[PubMed PMID: 25695963]
[28]
Kim W, Lee JE, Kim SH, Huh SY, Hyun JW, Jeong IH, Park MS, Cho JY, Lee SH, Lee KS, Kim HJ. Cerebral Cortex Involvement in Neuromyelitis Optica Spectrum Disorder. Journal of clinical neurology (Seoul, Korea). 2016 Apr:12(2):188-93. doi: 10.3988/jcn.2016.12.2.188. Epub 2016 Jan 28
[PubMed PMID: 26833983]
[29]
Kim SH, Kim W, Li XF, Jung IJ, Kim HJ. Does interferon beta treatment exacerbate neuromyelitis optica spectrum disorder? Multiple sclerosis (Houndmills, Basingstoke, England). 2012 Oct:18(10):1480-3
[PubMed PMID: 22354738]
[30]
Min JH, Kim BJ, Lee KH. Development of extensive brain lesions following fingolimod (FTY720) treatment in a patient with neuromyelitis optica spectrum disorder. Multiple sclerosis (Houndmills, Basingstoke, England). 2012 Jan:18(1):113-5. doi: 10.1177/1352458511431973. Epub 2011 Dec 6
[PubMed PMID: 22146605]
[31]
Jacob A, Hutchinson M, Elsone L, Kelly S, Ali R, Saukans I, Tubridy N, Boggild M. Does natalizumab therapy worsen neuromyelitis optica? Neurology. 2012 Sep 4:79(10):1065-6. doi: 10.1212/WNL.0b013e31826845fe. Epub 2012 Aug 22
[PubMed PMID: 22914835]
[32]
Dutra BG, da Rocha AJ, Nunes RH, Maia ACM Júnior. Neuromyelitis Optica Spectrum Disorders: Spectrum of MR Imaging Findings and Their Differential Diagnosis. Radiographics : a review publication of the Radiological Society of North America, Inc. 2018 Jan-Feb:38(1):169-193. doi: 10.1148/rg.2018170141. Epub
[PubMed PMID: 29320331]
[33]
Zalewski NL, Morris PP, Weinshenker BG, Lucchinetti CF, Guo Y, Pittock SJ, Krecke KN, Kaufmann TJ, Wingerchuk DM, Kumar N, Flanagan EP. Ring-enhancing spinal cord lesions in neuromyelitis optica spectrum disorders. Journal of neurology, neurosurgery, and psychiatry. 2017 Mar:88(3):218-225. doi: 10.1136/jnnp-2016-314738. Epub 2016 Dec 2
[PubMed PMID: 27913626]
[35]
Gómez-Figueroa E, Alvarado-Bolaños A, García-Estrada C, Zabala-Ángeles I, Sánchez-Rosales N, Bribiesca-Contreras E, García-Alvarez G, Montes-Pérez Y, Ramos-Vega E, Casallas-Vanegas A, Carrillo-Loza K, Corona-Vázquez T, Rivas-Alonso V, Flores-Rivera J. Clinical experience of plasmapheresis for neuromyelitis optica patients in Mexico. Multiple sclerosis and related disorders. 2021 Jul:52():103022. doi: 10.1016/j.msard.2021.103022. Epub 2021 May 15
[PubMed PMID: 34034213]
[36]
Li X, Tian DC, Fan M, Xiu Y, Wang X, Li T, Jia D, Xu W, Song T, Shi FD, Zhang X. Intravenous immunoglobulin for acute attacks in neuromyelitis optica spectrum disorders (NMOSD). Multiple sclerosis and related disorders. 2020 Sep:44():102325. doi: 10.1016/j.msard.2020.102325. Epub 2020 Jun 26
[PubMed PMID: 32653803]
[37]
Mealy MA, Wingerchuk DM, Palace J, Greenberg BM, Levy M. Comparison of relapse and treatment failure rates among patients with neuromyelitis optica: multicenter study of treatment efficacy. JAMA neurology. 2014 Mar:71(3):324-30. doi: 10.1001/jamaneurol.2013.5699. Epub
[PubMed PMID: 24445513]
Level 2 (mid-level) evidence
[38]
Agasing A, Quinn JL, Kumar G, Axtell RC. Interferon-β Intensifies Interleukin-23-Driven Pathogenicity of T Helper Cells in Neuroinflammatory Disease. Cells. 2021 Aug 20:10(8):. doi: 10.3390/cells10082139. Epub 2021 Aug 20
[PubMed PMID: 34440908]
[39]
Hegen H, Reindl M. Recent developments in MOG-IgG associated neurological disorders. Therapeutic advances in neurological disorders. 2020:13():1756286420945135. doi: 10.1177/1756286420945135. Epub 2020 Jul 31
[PubMed PMID: 33029200]
Level 3 (low-level) evidence
[40]
Huda S, Whittam D, Bhojak M, Chamberlain J, Noonan C, Jacob A. Neuromyelitis optica spectrum disorders. Clinical medicine (London, England). 2019 Mar:19(2):169-176. doi: 10.7861/clinmedicine.19-2-169. Epub
[PubMed PMID: 30872305]
[41]
Sellner J, Boggild M, Clanet M, Hintzen RQ, Illes Z, Montalban X, Du Pasquier RA, Polman CH, Sorensen PS, Hemmer B. EFNS guidelines on diagnosis and management of neuromyelitis optica. European journal of neurology. 2010 Aug:17(8):1019-32. doi: 10.1111/j.1468-1331.2010.03066.x. Epub 2010 Jun 7
[PubMed PMID: 20528913]