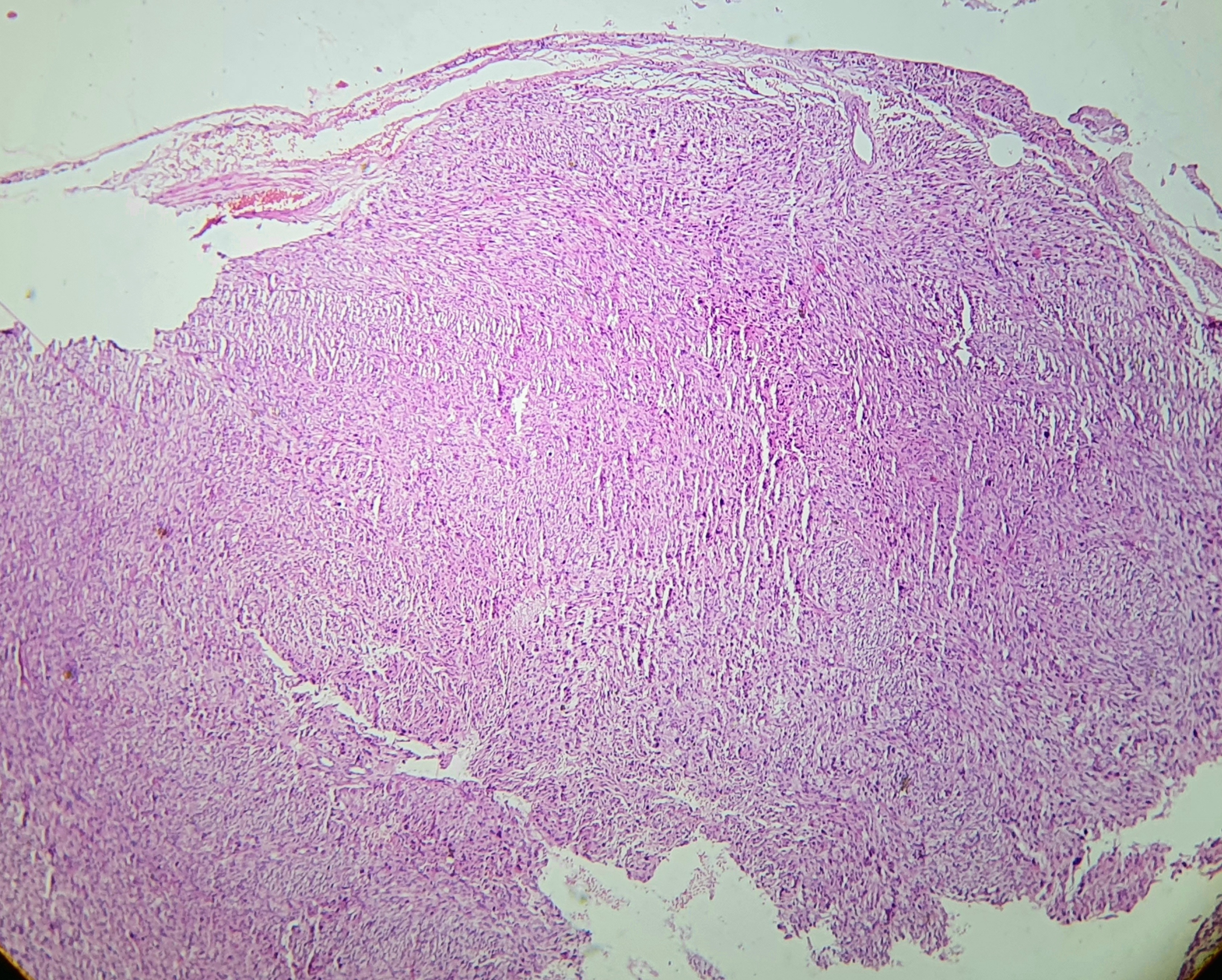
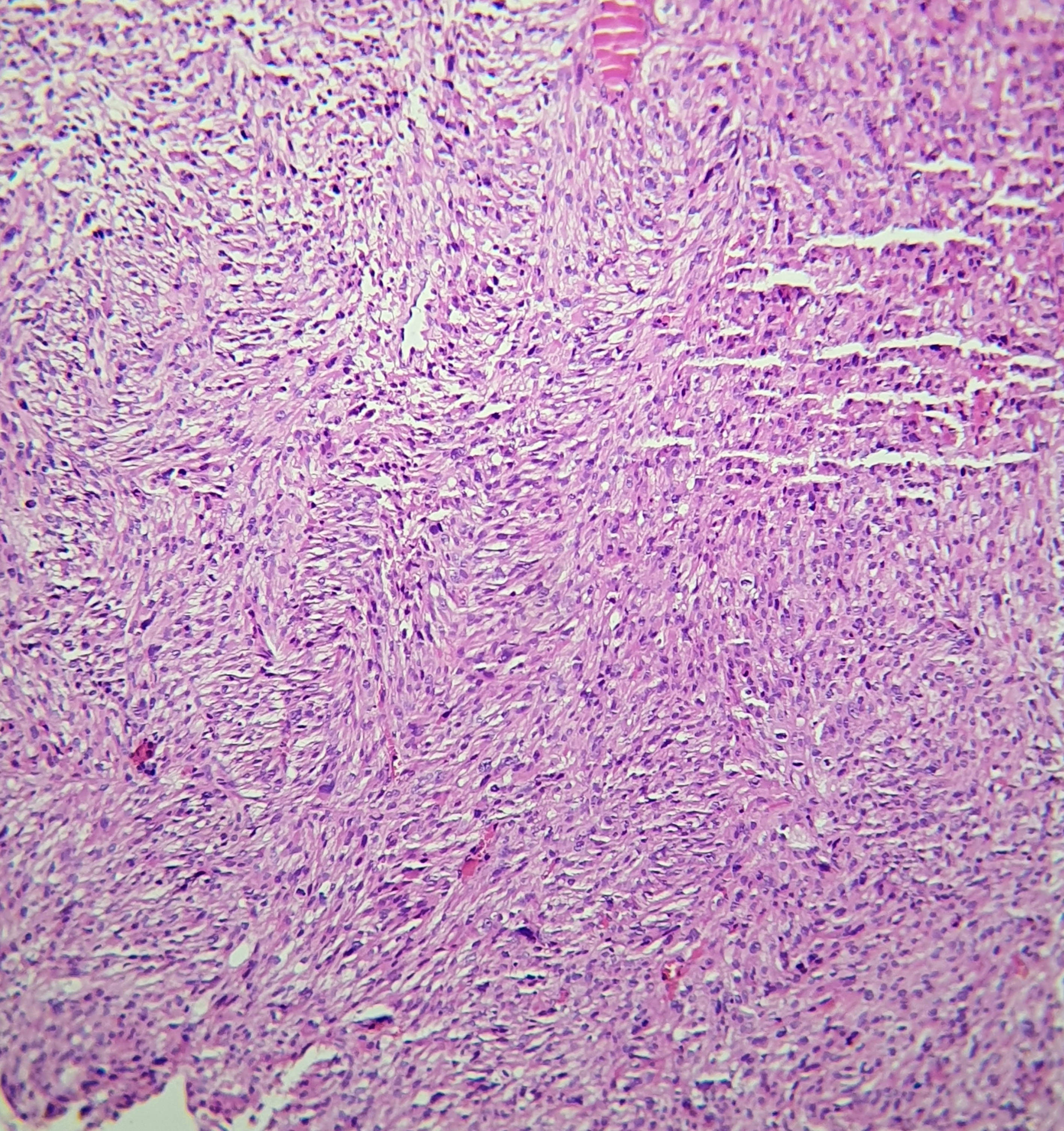
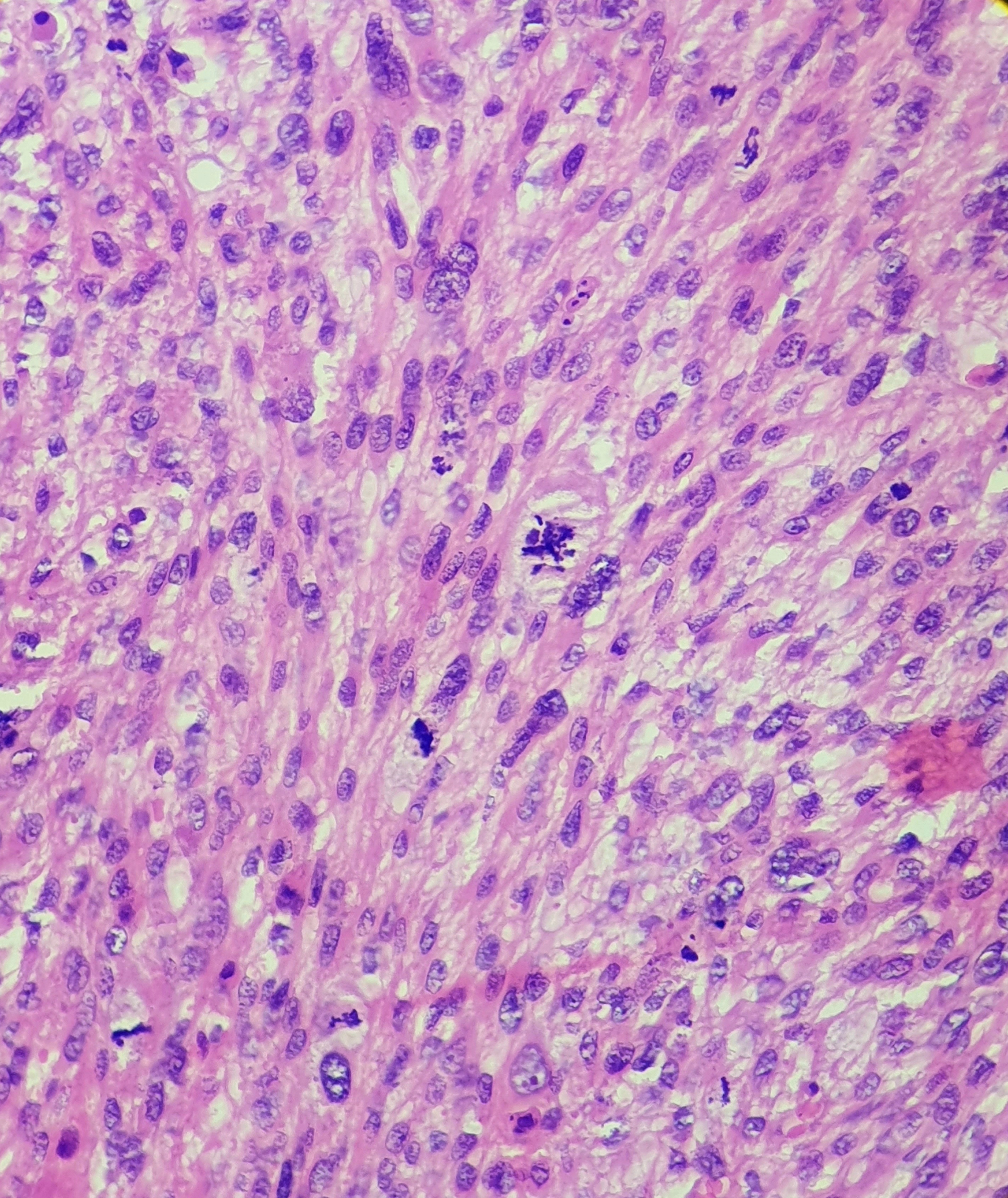
[1]
Goldblum JR. An approach to pleomorphic sarcomas: can we subclassify, and does it matter? Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2014 Jan:27 Suppl 1():S39-46. doi: 10.1038/modpathol.2013.174. Epub
[PubMed PMID: 24384852]
[2]
Weiss SW, Enzinger FM. Malignant fibrous histiocytoma: an analysis of 200 cases. Cancer. 1978 Jun:41(6):2250-66
[PubMed PMID: 207408]
Level 3 (low-level) evidence
[3]
Hornick JL. Cutaneous soft tissue tumors: how do we make sense of fibrous and "fibrohistiocytic" tumors with confusing names and similar appearances? Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2020 Jan:33(Suppl 1):56-65. doi: 10.1038/s41379-019-0388-4. Epub 2019 Oct 25
[PubMed PMID: 31653978]
[4]
Kelleher FC, Viterbo A. Histologic and genetic advances in refining the diagnosis of "undifferentiated pleomorphic sarcoma". Cancers. 2013 Feb 22:5(1):218-33. doi: 10.3390/cancers5010218. Epub 2013 Feb 22
[PubMed PMID: 24216705]
Level 3 (low-level) evidence
[5]
Widemann BC, Italiano A. Biology and Management of Undifferentiated Pleomorphic Sarcoma, Myxofibrosarcoma, and Malignant Peripheral Nerve Sheath Tumors: State of the Art and Perspectives. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2018 Jan 10:36(2):160-167. doi: 10.1200/JCO.2017.75.3467. Epub 2017 Dec 8
[PubMed PMID: 29220302]
Level 3 (low-level) evidence
[6]
Wang CY, Wei Q, Han I, Sato S, Ghanbari-Azarnier R, Whetstone H, Poon R, Hu J, Zheng F, Zhang P, Wang W, Wunder JS, Alman BA. Hedgehog and Notch signaling regulate self-renewal of undifferentiated pleomorphic sarcomas. Cancer research. 2012 Feb 15:72(4):1013-22. doi: 10.1158/0008-5472.CAN-11-2531. Epub 2012 Jan 9
[PubMed PMID: 22232736]
[7]
Hélias-Rodzewicz Z, Pérot G, Chibon F, Ferreira C, Lagarde P, Terrier P, Coindre JM, Aurias A. YAP1 and VGLL3, encoding two cofactors of TEAD transcription factors, are amplified and overexpressed in a subset of soft tissue sarcomas. Genes, chromosomes & cancer. 2010 Dec:49(12):1161-71. doi: 10.1002/gcc.20825. Epub
[PubMed PMID: 20842732]
[8]
Stefano S, Giovanni S. The PTEN Tumor Suppressor Gene in Soft Tissue Sarcoma. Cancers. 2019 Aug 14:11(8):. doi: 10.3390/cancers11081169. Epub 2019 Aug 14
[PubMed PMID: 31416195]
[9]
Matushansky I, Hernando E, Socci ND, Mills JE, Matos TA, Edgar MA, Singer S, Maki RG, Cordon-Cardo C. Derivation of sarcomas from mesenchymal stem cells via inactivation of the Wnt pathway. The Journal of clinical investigation. 2007 Nov:117(11):3248-57
[PubMed PMID: 17948129]
[10]
Pérot G, Chibon F, Montero A, Lagarde P, de Thé H, Terrier P, Guillou L, Ranchère D, Coindre JM, Aurias A. Constant p53 pathway inactivation in a large series of soft tissue sarcomas with complex genetics. The American journal of pathology. 2010 Oct:177(4):2080-90. doi: 10.2353/ajpath.2010.100104. Epub
[PubMed PMID: 20884963]
[11]
Simons A, Schepens M, Jeuken J, Sprenger S, van de Zande G, Bjerkehagen B, Forus A, Weibolt V, Molenaar I, van den Berg E, Myklebost O, Bridge J, van Kessel AG, Suijkerbuijk R. Frequent loss of 9p21 (p16(INK4A)) and other genomic imbalances in human malignant fibrous histiocytoma. Cancer genetics and cytogenetics. 2000 Apr 15:118(2):89-98
[PubMed PMID: 10748288]
[12]
Zheng B, Qu Y, Wang J, Shi Y, Yan W. Pathogenic and Targetable Genetic Alterations in Resected Recurrent Undifferentiated Pleomorphic Sarcomas Identified by Targeted Next-generation Sequencing. Cancer genomics & proteomics. 2019 May-Jun:16(3):221-228. doi: 10.21873/cgp.20127. Epub
[PubMed PMID: 31018952]
[13]
Hofvander J, Tayebwa J, Nilsson J, Magnusson L, Brosjö O, Larsson O, Vult von Steyern F, Mandahl N, Fletcher CD, Mertens F. Recurrent PRDM10 gene fusions in undifferentiated pleomorphic sarcoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2015 Feb 15:21(4):864-9. doi: 10.1158/1078-0432.CCR-14-2399. Epub 2014 Dec 16
[PubMed PMID: 25516889]
[14]
Delespaul L, Lesluyes T, Pérot G, Brulard C, Lartigue L, Baud J, Lagarde P, Le Guellec S, Neuville A, Terrier P, Vince-Ranchère D, Schmidt S, Debant A, Coindre JM, Chibon F. Recurrent TRIO Fusion in Nontranslocation-Related Sarcomas. Clinical cancer research : an official journal of the American Association for Cancer Research. 2017 Feb 1:23(3):857-867. doi: 10.1158/1078-0432.CCR-16-0290. Epub 2016 Aug 15
[PubMed PMID: 27528700]
[15]
Dineen SP, Roland CL, Feig R, May C, Zhou S, Demicco E, Sannaa GA, Ingram D, Wang WL, Ravi V, Guadagnolo A, Lev D, Pollock RE, Hunt K, Cormier J, Lazar A, Feig B, Torres KE. Radiation-Associated Undifferentiated Pleomorphic Sarcoma is Associated with Worse Clinical Outcomes than Sporadic Lesions. Annals of surgical oncology. 2015 Nov:22(12):3913-20. doi: 10.1245/s10434-015-4453-z. Epub 2015 Mar 6
[PubMed PMID: 25743327]
Level 2 (mid-level) evidence
[16]
Choi JH, Ro JY. The 2020 WHO Classification of Tumors of Soft Tissue: Selected Changes and New Entities. Advances in anatomic pathology. 2021 Jan:28(1):44-58. doi: 10.1097/PAP.0000000000000284. Epub
[PubMed PMID: 32960834]
Level 3 (low-level) evidence
[17]
Toro JR, Travis LB, Wu HJ, Zhu K, Fletcher CD, Devesa SS. Incidence patterns of soft tissue sarcomas, regardless of primary site, in the surveillance, epidemiology and end results program, 1978-2001: An analysis of 26,758 cases. International journal of cancer. 2006 Dec 15:119(12):2922-30
[PubMed PMID: 17013893]
Level 3 (low-level) evidence
[18]
von Mehren M, Kane JM, Bui MM, Choy E, Connelly M, Dry S, Ganjoo KN, George S, Gonzalez RJ, Heslin MJ, Homsi J, Keedy V, Kelly CM, Kim E, Liebner D, McCarter M, McGarry SV, Meyer C, Pappo AS, Parkes AM, Paz IB, Petersen IA, Poppe M, Riedel RF, Rubin B, Schuetze S, Shabason J, Sicklick JK, Spraker MB, Zimel M, Bergman MA, George GV. NCCN Guidelines Insights: Soft Tissue Sarcoma, Version 1.2021. Journal of the National Comprehensive Cancer Network : JNCCN. 2020 Dec 2:18(12):1604-1612. doi: 10.6004/jnccn.2020.0058. Epub 2020 Dec 2
[PubMed PMID: 33285515]
[19]
Nascimento AF, Raut CP. Diagnosis and management of pleomorphic sarcomas (so-called "MFH") in adults. Journal of surgical oncology. 2008 Mar 15:97(4):330-9. doi: 10.1002/jso.20972. Epub
[PubMed PMID: 18286476]
[20]
Rydholm A, Gustafson P. Should tumor depth be included in prognostication of soft tissue sarcoma? BMC cancer. 2003 May 26:3():17
[PubMed PMID: 12769830]
[21]
Henderson MT, Hollmig ST. Malignant fibrous histiocytoma: changing perceptions and management challenges. Journal of the American Academy of Dermatology. 2012 Dec:67(6):1335-41. doi: 10.1016/j.jaad.2012.04.013. Epub 2012 Jun 5
[PubMed PMID: 22677489]
[22]
Hornick JL. Subclassification of pleomorphic sarcomas: How and why should we care? Annals of diagnostic pathology. 2018 Dec:37():118-124. doi: 10.1016/j.anndiagpath.2018.10.006. Epub 2018 Oct 11
[PubMed PMID: 30340082]
[23]
Lazova R, Moynes R, May D, Scott G. LN-2 (CD74). A marker to distinguish atypical fibroxanthoma from malignant fibrous histiocytoma. Cancer. 1997 Jun 1:79(11):2115-24
[PubMed PMID: 9179057]
[24]
Leader M, Collins M, Patel J, Henry K. Vimentin: an evaluation of its role as a tumour marker. Histopathology. 1987 Jan:11(1):63-72
[PubMed PMID: 2435649]
[25]
Roland CL, May CD, Watson KL, Al Sannaa GA, Dineen SP, Feig R, Landers S, Ingram DR, Wang WL, Guadagnolo BA, Feig B, Hunt KK, Cormier JN, Lazar AJ, Torres KE. Analysis of Clinical and Molecular Factors Impacting Oncologic Outcomes in Undifferentiated Pleomorphic Sarcoma. Annals of surgical oncology. 2016 Jul:23(7):2220-8. doi: 10.1245/s10434-016-5115-5. Epub 2016 Feb 3
[PubMed PMID: 26847678]
[26]
Hanlon A, Stasko T, Christiansen D, Cyrus N, Galan A. LN2, CD10, and Ezrin Do Not Distinguish Between Atypical Fibroxanthoma and Undifferentiated Pleomorphic Sarcoma or Predict Clinical Outcome. Dermatologic surgery : official publication for American Society for Dermatologic Surgery [et al.]. 2017 Mar:43(3):431-436. doi: 10.1097/DSS.0000000000001000. Epub
[PubMed PMID: 28079637]
Level 2 (mid-level) evidence
[27]
Povar J, Franco JM, Muñoz JR, Horndler C, Arazo P, Portoles A, Velilla J, Gutiérrez A. [Malignant fibrous histiocytoma of soft tissue: description of 10 cases]. Medicina clinica. 1991 Jan 12:96(1):6-10
[PubMed PMID: 1850813]
Level 3 (low-level) evidence
[28]
Kawarabayashi T, Okuno K, Niki K, Nakata T, Matsumoto M, Otani S, Wakami S, Yoshihara W, Taki T, Kaneda K, Nishiwaki N, Tane K. Primary cardiac malignant fibrous histiocytoma with abdominal wall metastasis. Journal of cardiology cases. 2015 Nov:12(5):139-142. doi: 10.1016/j.jccase.2015.05.014. Epub 2015 Jun 25
[PubMed PMID: 30546578]
Level 3 (low-level) evidence
[29]
Salem J, Shamseddine A, Khalife M, Nounou GE, El Naaj AA, Mukherji D, Haydar A, Faraj W. Malignant fibrous histiocytoma presenting with complete opacification of the hemithorax: A case report. International journal of surgery case reports. 2014:5(12):1162-3. doi: 10.1016/j.ijscr.2014.11.006. Epub 2014 Nov 11
[PubMed PMID: 25437664]
Level 3 (low-level) evidence
[30]
Chen S, Huang W, Luo P, Cai W, Yang L, Sun Z, Zheng B, Yan W, Wang C. Undifferentiated Pleomorphic Sarcoma: Long-Term Follow-Up from a Large Institution. Cancer management and research. 2019:11():10001-10009. doi: 10.2147/CMAR.S226896. Epub 2019 Nov 27
[PubMed PMID: 31819633]
[31]
Vodanovich DA, Spelman T, May D, Slavin J, Choong PFM. Predicting the prognosis of undifferentiated pleomorphic soft tissue sarcoma: a 20-year experience of 266 cases. ANZ journal of surgery. 2019 Sep:89(9):1045-1050. doi: 10.1111/ans.15348. Epub 2019 Jul 30
[PubMed PMID: 31364245]
Level 3 (low-level) evidence
[32]
Kubo T, Furuta T, Johan MP, Ochi M. Prognostic significance of (18)F-FDG PET at diagnosis in patients with soft tissue sarcoma and bone sarcoma; systematic review and meta-analysis. European journal of cancer (Oxford, England : 1990). 2016 May:58():104-11. doi: 10.1016/j.ejca.2016.02.007. Epub 2016 Mar 15
[PubMed PMID: 26990930]
Level 1 (high-level) evidence
[33]
Brown MD, Swanson NA. Treatment of malignant fibrous histiocytoma and atypical fibrous xanthomas with micrographic surgery. The Journal of dermatologic surgery and oncology. 1989 Dec:15(12):1287-92
[PubMed PMID: 2556464]
[34]
Huether MJ, Zitelli JA, Brodland DG. Mohs micrographic surgery for the treatment of spindle cell tumors of the skin. Journal of the American Academy of Dermatology. 2001 Apr:44(4):656-9
[PubMed PMID: 11260542]
[35]
Rosenberg SA, Tepper J, Glatstein E, Costa J, Baker A, Brennan M, DeMoss EV, Seipp C, Sindelar WF, Sugarbaker P, Wesley R. The treatment of soft-tissue sarcomas of the extremities: prospective randomized evaluations of (1) limb-sparing surgery plus radiation therapy compared with amputation and (2) the role of adjuvant chemotherapy. Annals of surgery. 1982 Sep:196(3):305-15
[PubMed PMID: 7114936]
Level 1 (high-level) evidence
[36]
Gronchi A, Ferrari S, Quagliuolo V, Broto JM, Pousa AL, Grignani G, Basso U, Blay JY, Tendero O, Beveridge RD, Ferraresi V, Lugowska I, Merlo DF, Fontana V, Marchesi E, Donati DM, Palassini E, Palmerini E, De Sanctis R, Morosi C, Stacchiotti S, Bagué S, Coindre JM, Dei Tos AP, Picci P, Bruzzi P, Casali PG. Histotype-tailored neoadjuvant chemotherapy versus standard chemotherapy in patients with high-risk soft-tissue sarcomas (ISG-STS 1001): an international, open-label, randomised, controlled, phase 3, multicentre trial. The Lancet. Oncology. 2017 Jun:18(6):812-822. doi: 10.1016/S1470-2045(17)30334-0. Epub 2017 May 9
[PubMed PMID: 28499583]
Level 1 (high-level) evidence
[37]
Gronchi A, Palmerini E, Quagliuolo V, Martin Broto J, Lopez Pousa A, Grignani G, Brunello A, Blay JY, Tendero O, Diaz Beveridge R, Ferraresi V, Lugowska I, Merlo DF, Fontana V, Marchesi E, Braglia L, Donati DM, Palassini E, Bianchi G, Marrari A, Morosi C, Stacchiotti S, Bagué S, Coindre JM, Dei Tos AP, Picci P, Bruzzi P, Casali PG. Neoadjuvant Chemotherapy in High-Risk Soft Tissue Sarcomas: Final Results of a Randomized Trial From Italian (ISG), Spanish (GEIS), French (FSG), and Polish (PSG) Sarcoma Groups. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2020 Jul 1:38(19):2178-2186. doi: 10.1200/JCO.19.03289. Epub 2020 May 18
[PubMed PMID: 32421444]
Level 1 (high-level) evidence
[38]
Keung EZ, Lazar AJ, Torres KE, Wang WL, Cormier JN, Ashleigh Guadagnolo B, Bishop AJ, Lin H, Hunt KK, Bird J, Lewis VO, Patel SR, Wargo JA, Somaiah N, Roland CL. Phase II study of neoadjuvant checkpoint blockade in patients with surgically resectable undifferentiated pleomorphic sarcoma and dedifferentiated liposarcoma. BMC cancer. 2018 Sep 24:18(1):913. doi: 10.1186/s12885-018-4829-0. Epub 2018 Sep 24
[PubMed PMID: 30249211]
[39]
Wisdom AJ, Mowery YM, Riedel RF, Kirsch DG. Rationale and emerging strategies for immune checkpoint blockade in soft tissue sarcoma. Cancer. 2018 Oct 1:124(19):3819-3829. doi: 10.1002/cncr.31517. Epub 2018 May 3
[PubMed PMID: 29723407]
[40]
Cates JMM. The AJCC 8th Edition Staging System for Soft Tissue Sarcoma of the Extremities or Trunk: A Cohort Study of the SEER Database. Journal of the National Comprehensive Cancer Network : JNCCN. 2018 Feb:16(2):144-152. doi: 10.6004/jnccn.2017.7042. Epub
[PubMed PMID: 29439175]
[41]
Winchester D, Lehman J, Tello T, Chimato N, Hocker T, Kim S, Chang J, Markey J, Yom SS, Ryan W, Mully T, Hodge D, Otley C, Arron ST. Undifferentiated pleomorphic sarcoma: Factors predictive of adverse outcomes. Journal of the American Academy of Dermatology. 2018 Nov:79(5):853-859. doi: 10.1016/j.jaad.2018.05.022. Epub 2018 May 19
[PubMed PMID: 29787841]
[42]
Abouarab MH, Salem IL, Degheidy MM, Henn D, Hirche C, Eweida A, Uhl M, Kneser U, Kremer T. Therapeutic options and postoperative wound complications after extremity soft tissue sarcoma resection and postoperative external beam radiotherapy. International wound journal. 2018 Feb:15(1):148-158. doi: 10.1111/iwj.12851. Epub 2017 Dec 5
[PubMed PMID: 29205902]
[43]
Elswick SM, Curiel DA, Wu P, Akhavan A, Molinar VE, Mohan AT, Sim FH, Martinez-Jorge J, Saint-Cyr M. Complications after thigh sarcoma resection. Journal of surgical oncology. 2020 May:121(6):945-951. doi: 10.1002/jso.25830. Epub 2020 Feb 4
[PubMed PMID: 32020627]
[44]
Srikanthan A, Leung B, Shokoohi A, Smrke A, Bates A, Ho C. Psychosocial Distress Scores and Needs among Newly Diagnosed Sarcoma Patients: A Provincial Experience. Sarcoma. 2019:2019():5302639. doi: 10.1155/2019/5302639. Epub 2019 Jul 1
[PubMed PMID: 31354383]
[45]
Weaver R, O'Connor M, Sobhi S, Carey Smith R, Halkett G. The unmet needs of patients with sarcoma. Psycho-oncology. 2020 Jul:29(7):1209-1216. doi: 10.1002/pon.5411. Epub 2020 Jun 16
[PubMed PMID: 32419264]
[46]
Smith SR. Rehabilitation Strategies and Outcomes of the Sarcoma Patient. Physical medicine and rehabilitation clinics of North America. 2017 Feb:28(1):171-180. doi: 10.1016/j.pmr.2016.08.008. Epub
[PubMed PMID: 27912995]